|
|
Texto e ilustrações com links para casos |
|
|
|
Texto e ilustrações com links para casos |
|
| NEUROFIBROMATOSE
DO TIPO 1. (NF1)
Facomatoses. NF1 é uma das facomatoses, que são malformações congênitas afetando principalmente estruturas de origem ectodérmica: o sistema nervoso, globo ocular e pele. Classicamente, estão incluídas no grupo quatro doenças: a NF1, esclerose tuberosa ou doença de Bourneville, angiomatose retino-cerebelar ou doença de von HippelLindau, e angiomatose encéfalo-trigeminal ou síndrome de Sturge-Weber. NF1, descrita por von Recklinghausen em 1882, é também conhecida como neurofibromatose de Von Recklinghausen ou periférica. (Contudo, é melhor evitar os termos periférico e central em relação às neurofibromatoses, pois tanto a NF1 tem freqüentemente lesões centrais como a NF2 pode ocasionalmente ter lesões periféricas). Cerca de 15 a 20% dos pacientes com NF1 têm manifestações envolvendo o SNC. Definição. NF1 é uma doença autossômica dominante caracterizada por múltiplos neurofibromas, neurofibromas plexiformes, que podem evoluir para o tumor maligno da bainha de nervos periféricos, gliomas de nervo óptico e outros astrocitomas, lesões melânicas, como manchas café com leite, sardas axilares e inguinais e hamartomas da íris (nódulos de Lisch) e várias lesões ósseas. O risco de desenvolver neoplasias é 4 vezes maior em um paciente com NF1 do que na população geral. Incidência 1:4000 na população geral (uma das doenças genéticas mais comuns). Cerca de metade dos casos é por novas mutações. A maioria das novas mutações ocorre na linha germinativa paterna. Há alta penetrância (quase todos pacientes portadores do gene manifestam a doença) mas a expressividade é altamente variável. Não há correlação entre o tipo de mutação e o quadro clínico. Critérios diagnósticos: dois ou mais dos seguintes: 1.
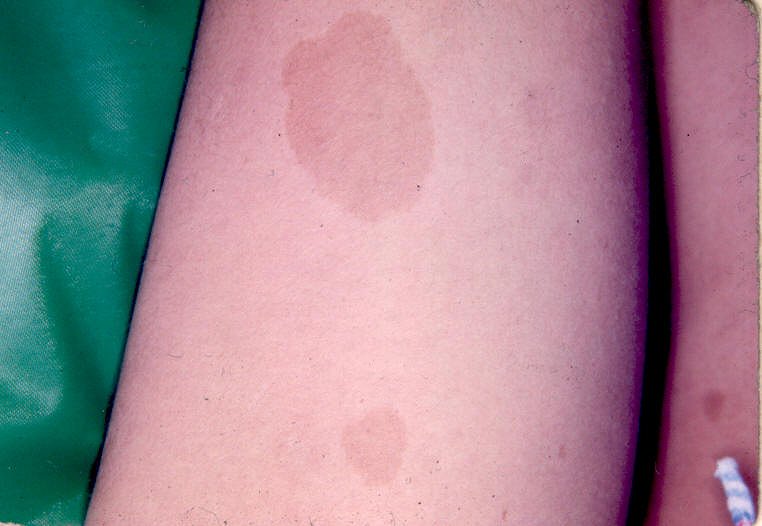
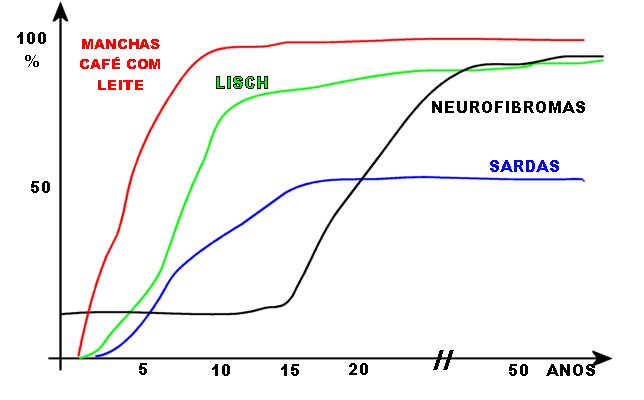
6 ou + manchas café com leite com diâmetro maior que 5 mm
em indivíduos pré-púberes e maior que 15 mm em indivíduos
pós-púberes;
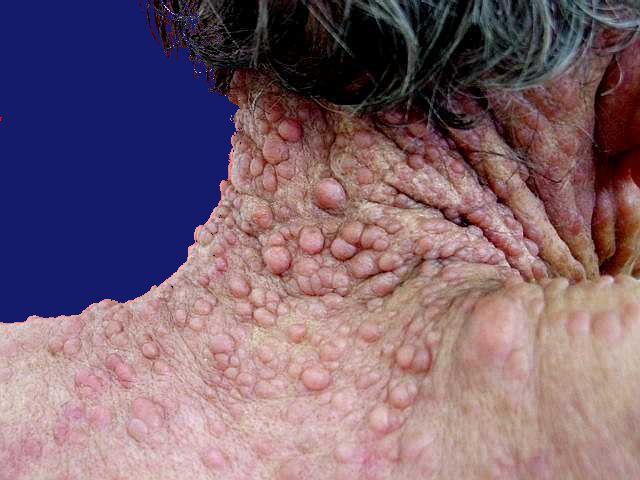
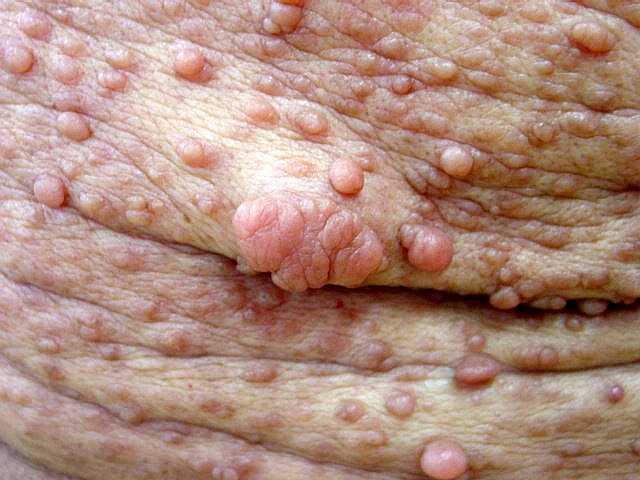
Anormalidades da pigmentação (i. e., dos melanócitos). Manchas café com leite aparecem geralmente durante o 1º ano de vida e são freqüentemente a primeira manifestação na doença no RN. Aumentam com a idade, chegando a > 95% dos pacientes, mas podem estabilizar ou regredir na idade adulta. Há um aumento da proporção de melanócitos / queratinócitos. Sardas axilares aparecem mais tarde, chegando a 65 a 85% dos pacientes, com maior prevalência em adultos jovens. Nódulos de Lisch (hamartomas pigmentados e e elevados na superfície da íris) melhor pesquisados com lâmpada de fenda. Começam a aparecer na infância e estão presentes em quase todos adultos afetados (0-4 anos 20%; 5-9 anos 40%; 10-19 anos 80%; acima dos 20 anos 95%). Por isso, são úteis para o diagnóstico. Neurofibromas cutâneos começam a aparecer no início da puberdade e aumentam em número ao longo da vida (0-9 anos 15%; 10-19 anos 45%; 20-29 anos 85%; acima dos 30 anos 95%). Neurofibromas plexiformes.
Encontrados em 1/4 a 1/3 dos casos. Limitados a cabeça e pescoço
em 2-4%. Malignização para tumor maligno de baínha
de nervos periféricos (malignant peripheral nerve sheath tumor
ou
MPNST) 2-3%.
Manifestações
relacionadas ao SNC.
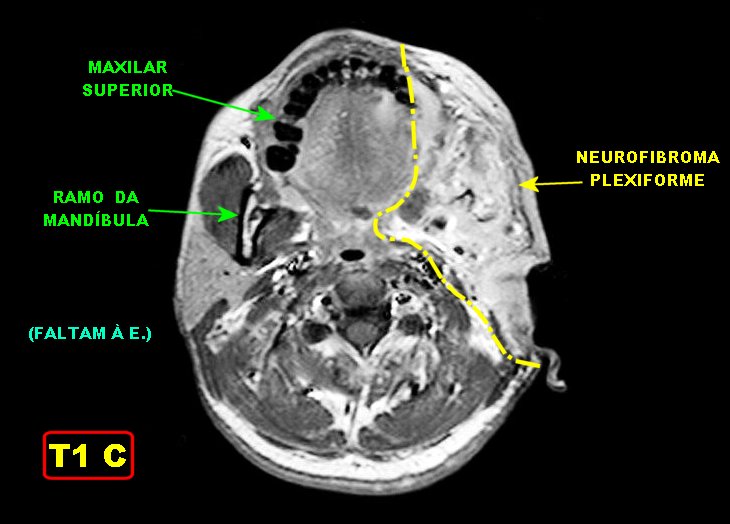
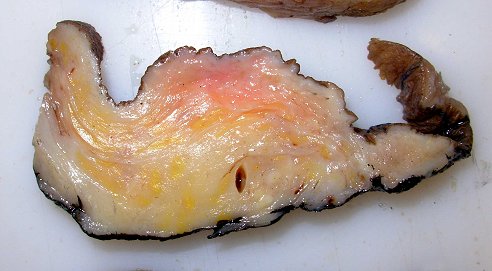
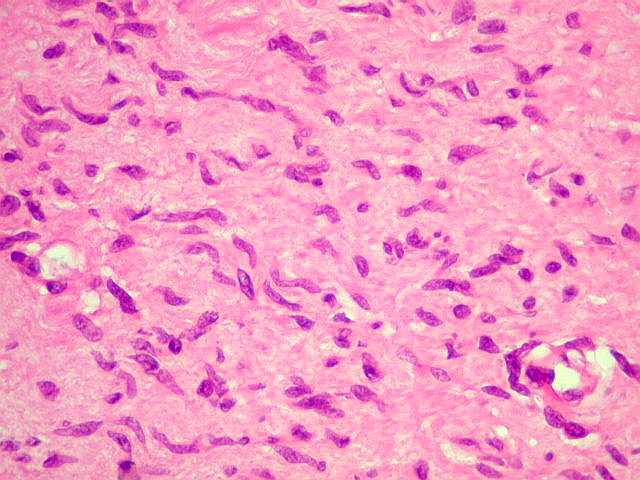
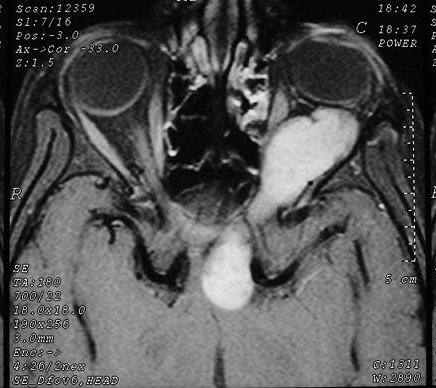
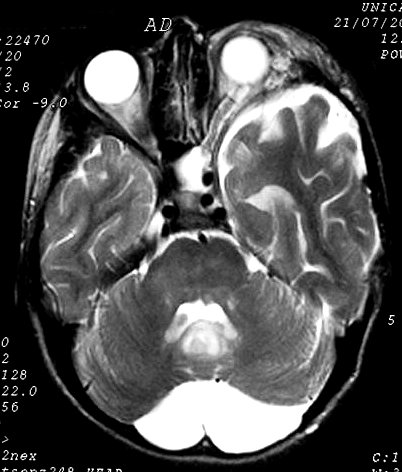
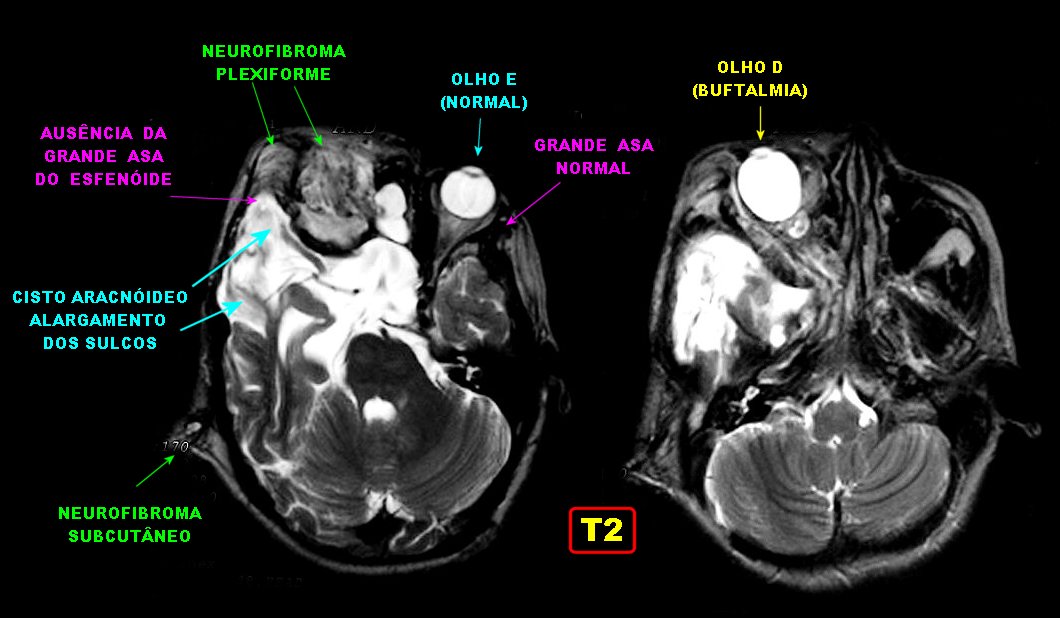
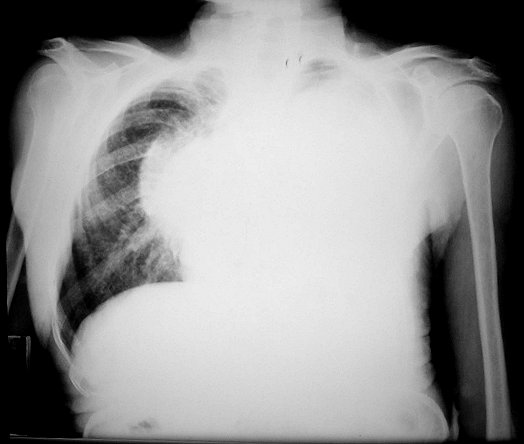
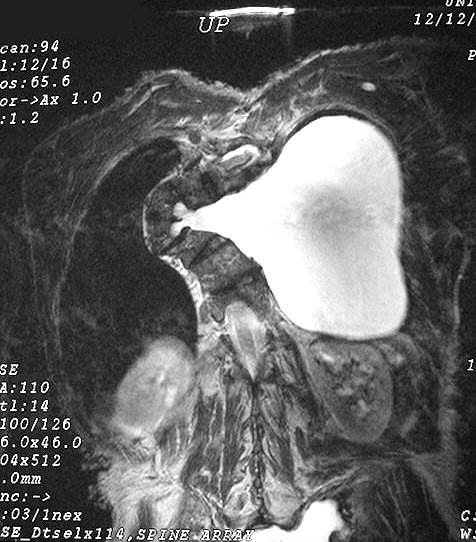
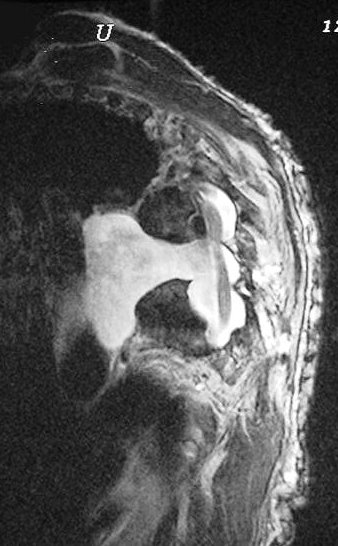
Anormalidades do esqueleto. Escoliose 15%; baixa estatura 30%; macrocefalia - 50%; pseudoartrose de ossos longos 3%; displasia da asa do esfenóide 1%. Outros estenose da A. renal, levando a hipertensão 1%. Manifestações clínicas, radiológicas, anátomo-patológicas na NF1 (detalhes). Os neurofibromas plexiformes
são praticamente patognomônicos da doença e
ocorrem em um quarto a um terço dos pacientes. Na cabeça
e pescoço, localizam-se preferencialmente na divisão orbitária
do trigêmeo (V1). Estão associados com displasia da asa do
esfenóide e/ou cistos aracnóideos de fossa média.
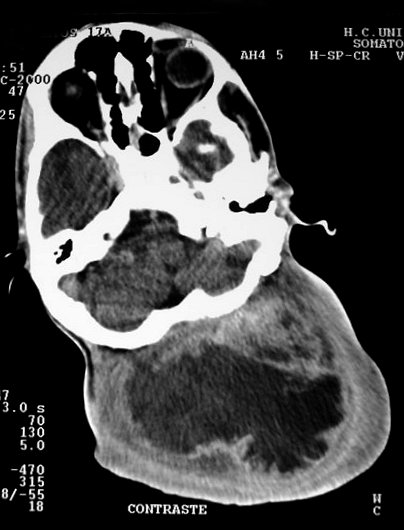
Em TC/RM aparecem como massas mal delimitadas no espaço mastigador
profundo e geralmente alcançam a órbita e seio cavernoso.
São isointensos ao músculo em T1 e se reforçam fortemente
com contraste (ao contrário dos músculos; só musculatura
extrínseca do globo ocular se impregna por contraste). Neurofibromas
plexiformes podem desenvolver-se já no primeiro ou segundo ano de
vida como um crescimento subcutâneo com margens mal definidas, e
podem causar deformidades grosseiras mais tardiamente, afetando extensas
áreas do corpo.
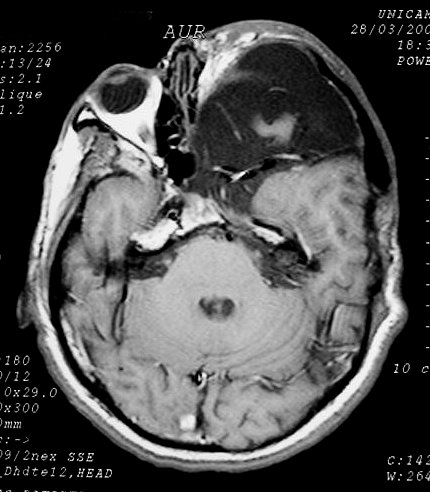
Neurofibromas malignos, neurofibrossarcomas ou MPNST ocorrem na NF1 com incidência de 3 a 13%, menor em crianças e aumentando com a idade. Aparecem como massas grandes e circunscritas, com variável invasão de estruturas próximas. São internamente heterogêneos, e assim também a impregnação por contraste. Os MPNST que ocorrem em NF1 aparecem em idade mais precoce e podem incluir elementos rabdomioblásticos (rabdomiossarcoma) e de outros tipos, inclusive glandulares. São chamados Triton tumors, e altamente característicos da doença. Na maioria
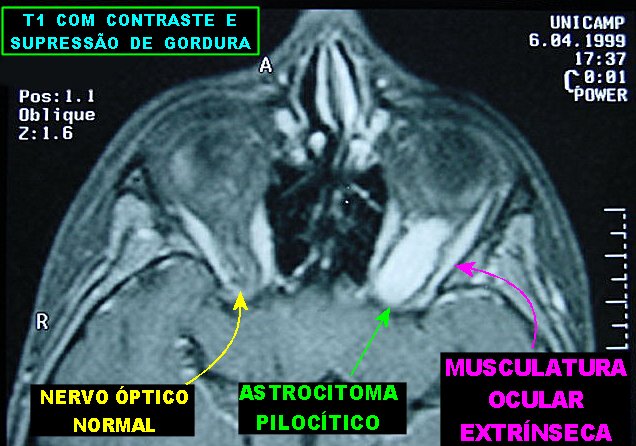
são astrocitomas pilocíticos do nervo óptico.
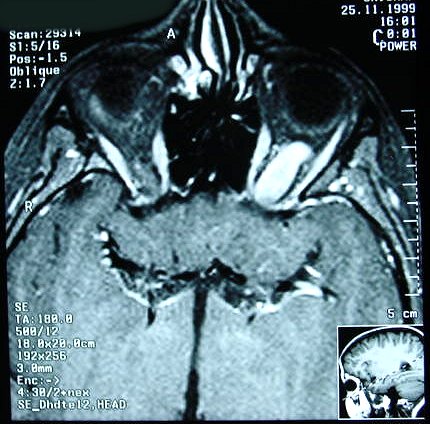
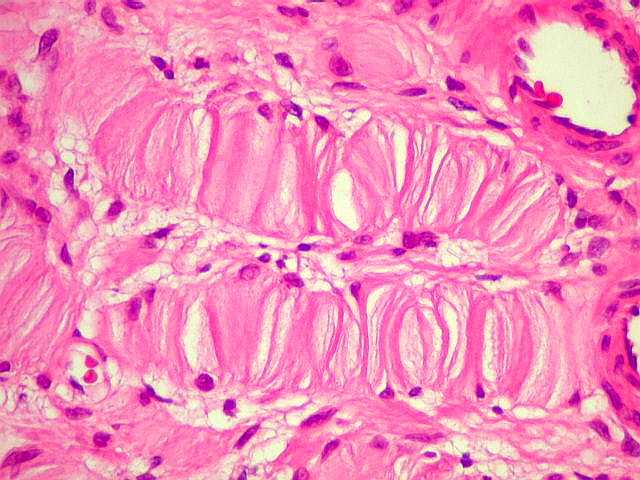
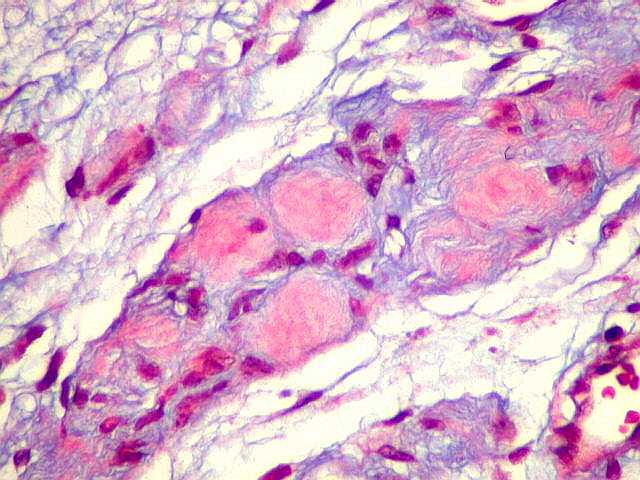
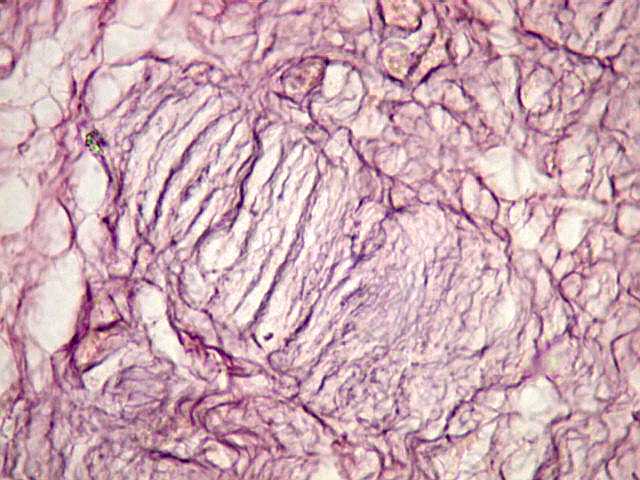
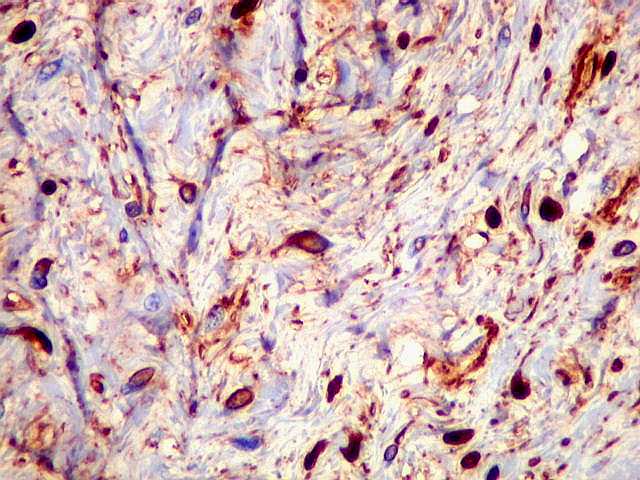
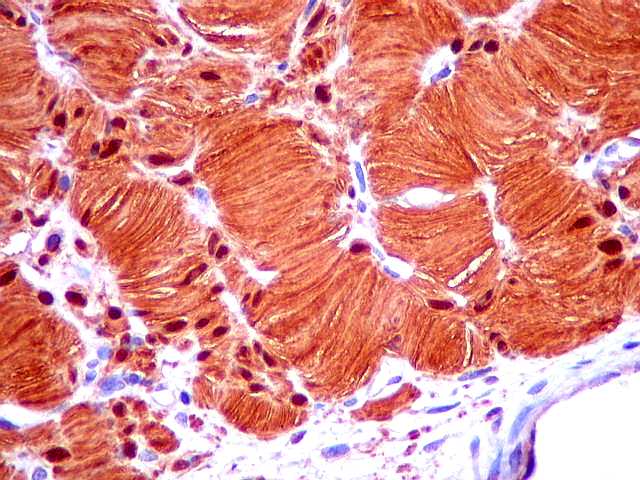
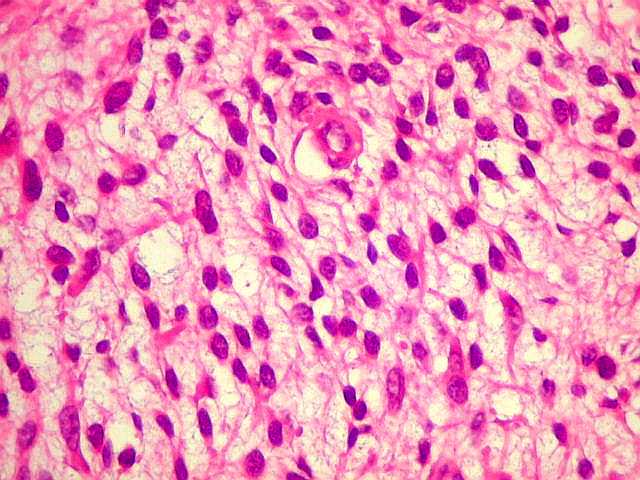
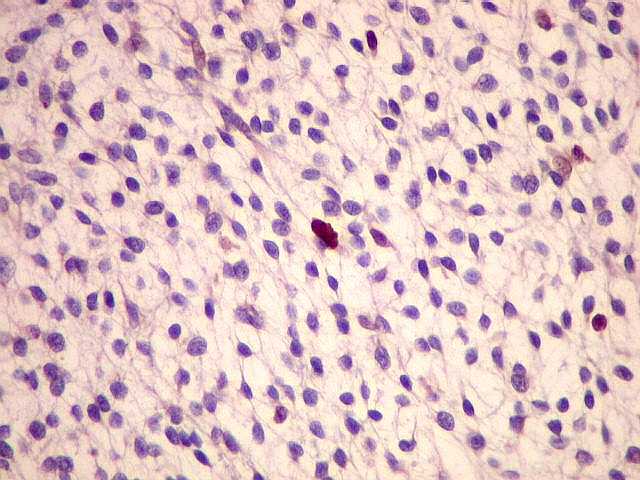
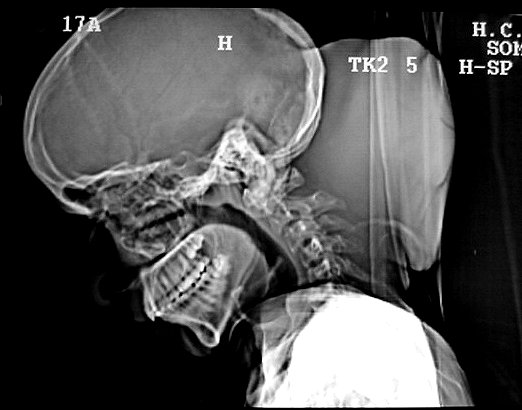
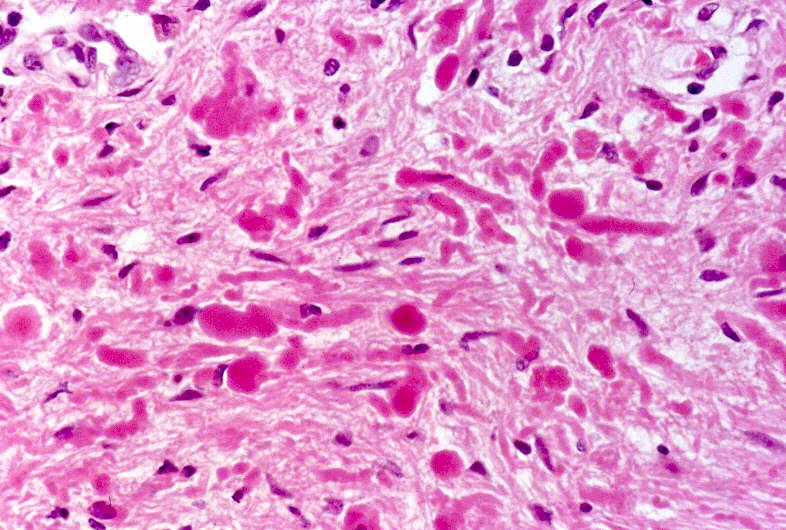
Ver casos de neuroimagem (1) (2),
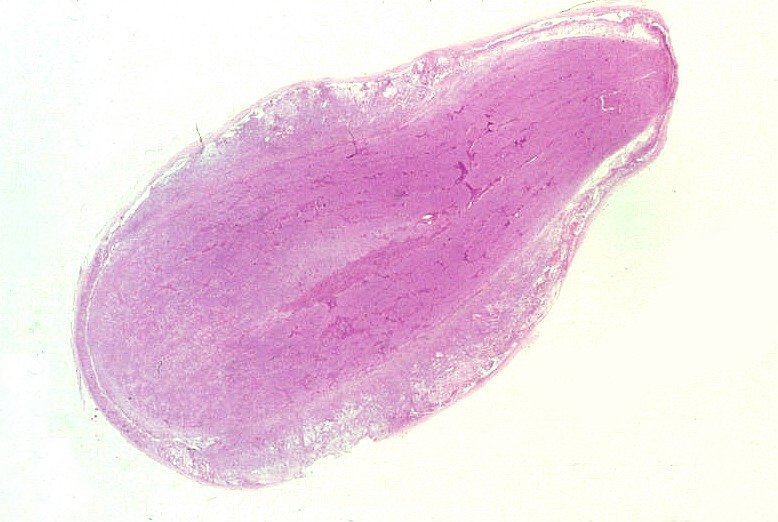
banco
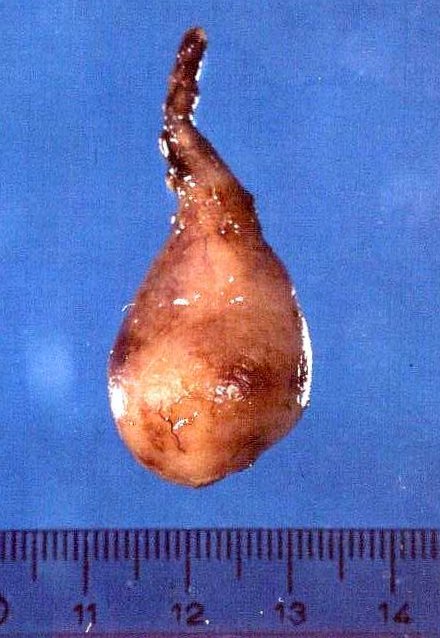
de imagens, e aspecto histopatológico.
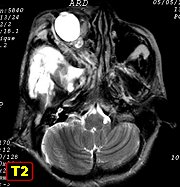
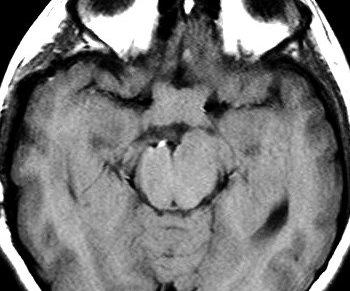
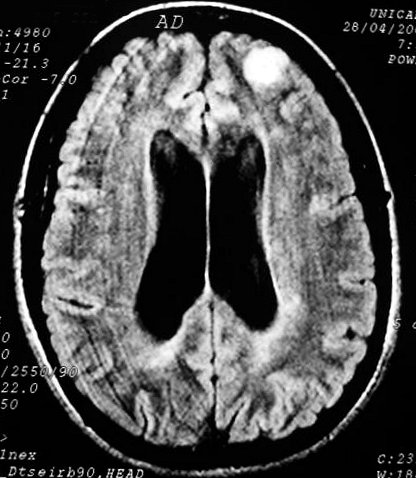
São a anormalidade cerebral primária mais comum na NF1 (15%),
mas só metade ou menos dos pacientes apresentam sintomas. Podem
ocorrer em um ou ambos nervos ópticos, quiasma e tratos ópticos.
Tumores restritos aos nervos ópticos podem ser assintomáticos
ou apresentar déficit visual que só raramente é progressivo.
Tumores envolvendo o quiasma e tratos ópticos têm pior prognóstico.
Tumores envolvendo o hipotálamo podem associar-se a puberdade precoce
(ver caso). Na grande maioria, os gliomas
ópticos progridem muito lentamente, são estáveis ou
podem regredir espontaneamente. Raramente, podem ser agressivos.
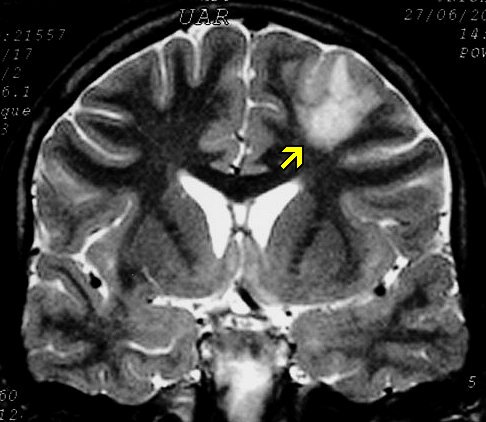
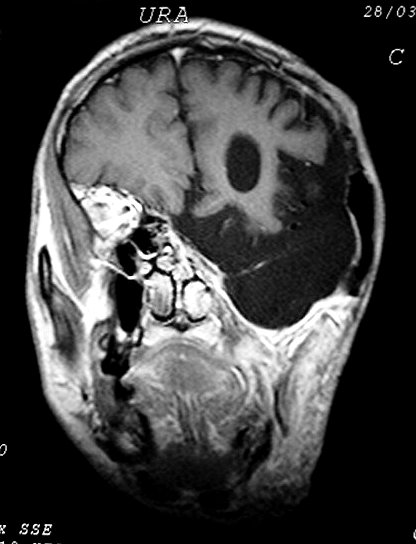
Outros astrocitomas.
São mais comuns na NF1 que na população em geral e
correspondem a astrocitomas difusos de baixo ou alto grau. As vias ópticas
são o local mais envolvido, mas há também preferência
pelo tronco cerebral. Os astrocitomas de tronco na NF1 ocorrem mais no
bulbo e mesencéfalo (principalmente na região
periaquedutal), e nisto diferem dos da população em geral,
que são mais comuns na ponte. Também têm curso mais
indolente e melhor prognóstico. Tumores do teto mesencefálico
podem até regredir espontaneamente. Abaixo, exemplos de astrocitoma
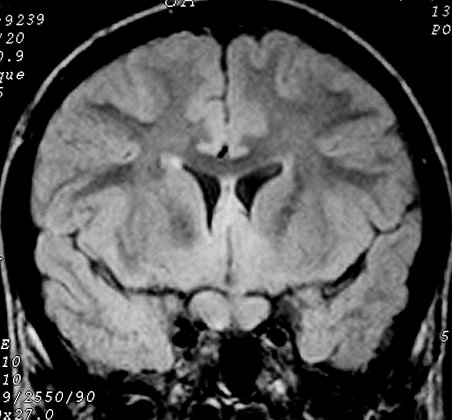
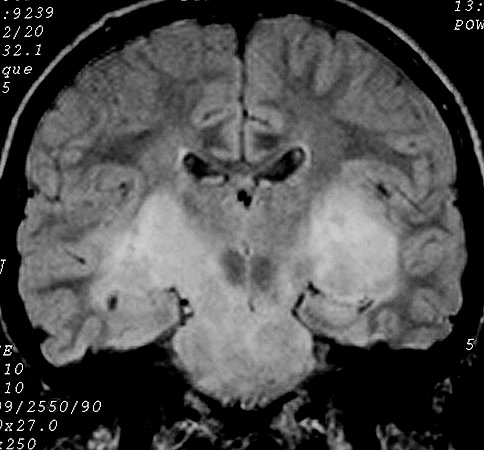
difuso das vias ópticas associado a disseminação tumoral
pelos hemisférios cerebrais (gliomatose).
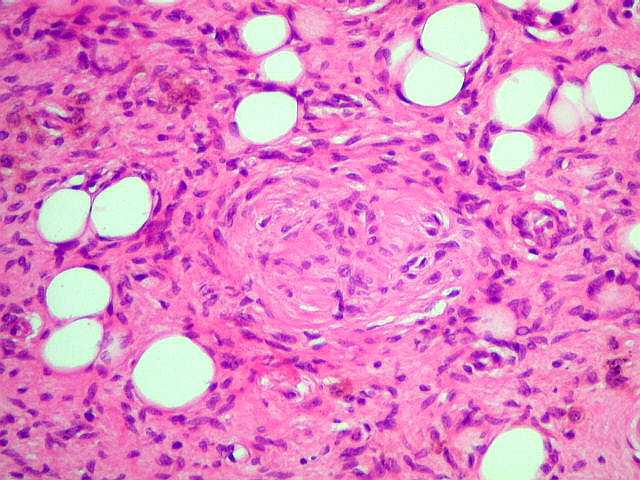
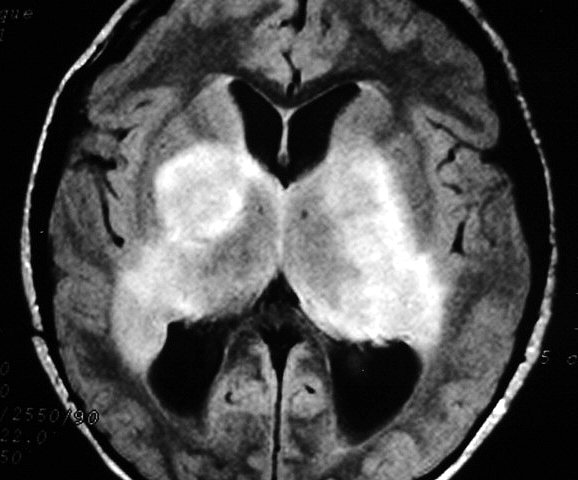
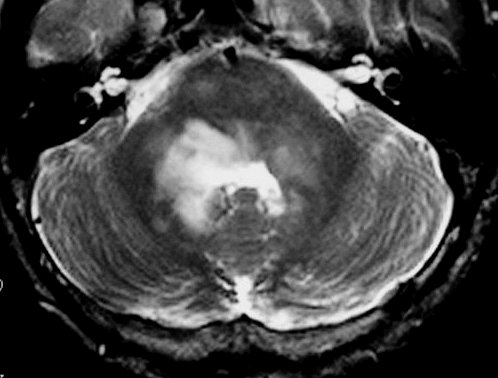
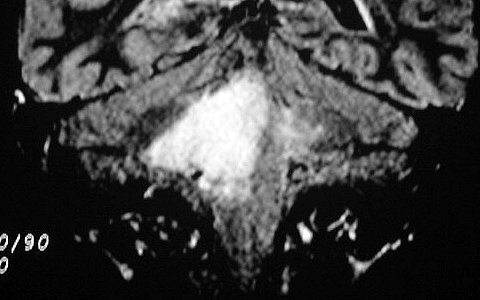
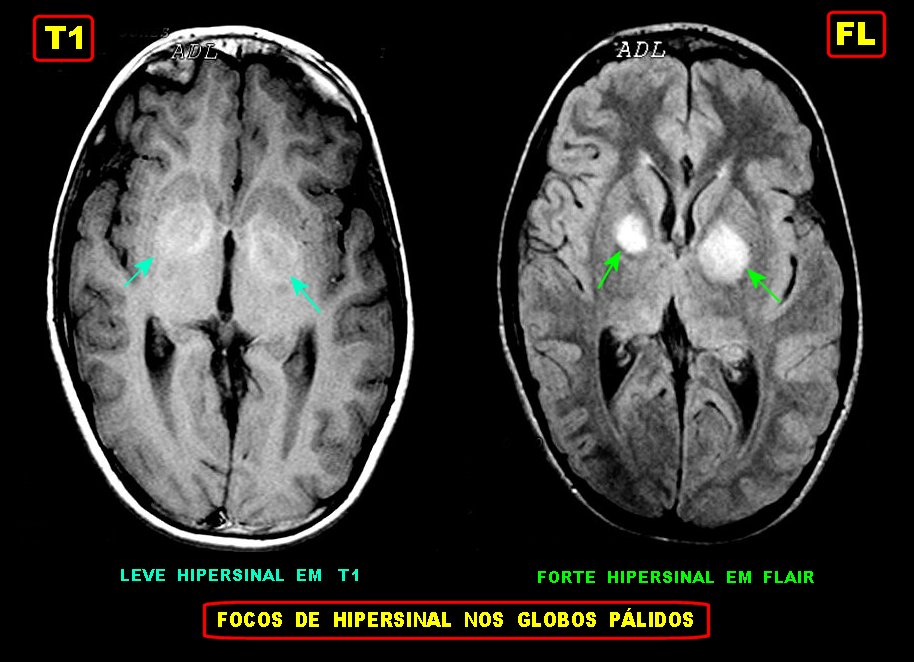
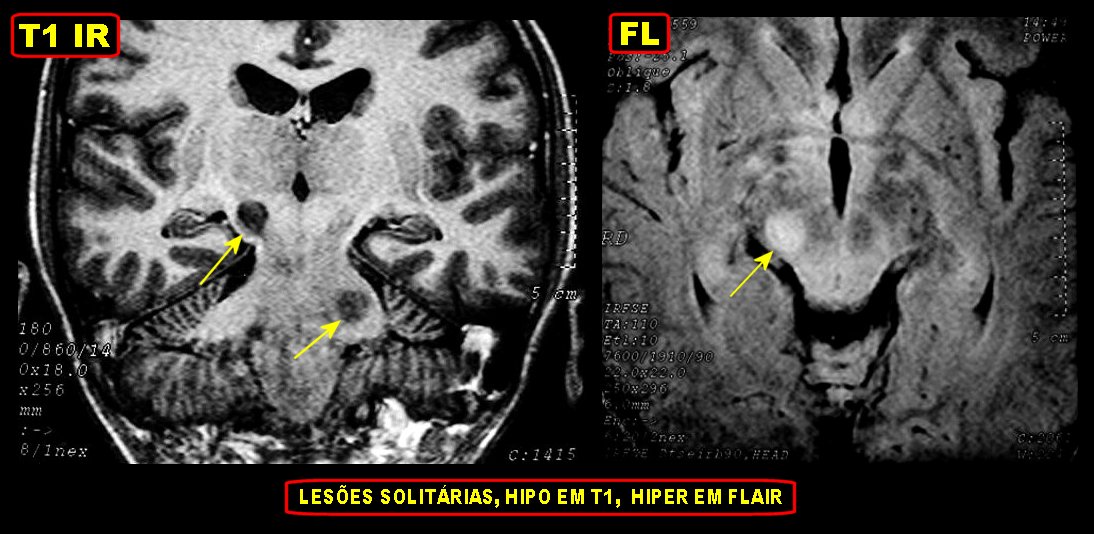
Tumores de nervos cranianos (exceto o N. óptico) são raros na NF1, que se caracteriza por tumores gliais (centrais) e neurofibromas de nervos periféricos. Schwannomas são muito mais comuns na NF2. Se ocorrerem schwannomas em paciente com NF1 deve-se levantar a possibilidade de síndromes de superposição (overlap syndromes). Focos de hipersinal no TR longo. São anormalidades não neoplásicas no parênquima cerebral, às vezes também referidas como lesões hamartomatosas .
Uma regra geral importante em NF1 é que o comportamento biológico de lesões da substância branca não pode ser previsto apenas com o aspecto em um exame. Lesões que parecem benignas podem progredir a neoplasia franca e lesões de aspecto neoplásico podem regredir ou desaparecer.
A dificuldade no aprendizado é definida como uma discrepância entre a habilidade (intelecto ou aptidão) e o desempenho, em pacientes com QI normal. Estimativas vão de 30 a 45% em crianças com NF1, ou seja 3 vezes mais que a população geral. Há uma distribuição bimodal do QI em pacientes com NF1, ou seja, os normais e os com déficit cognitivo. O déficit cognitivo parece estar associado aos focos de hipersinal em T2 (hamartomas) nos núcleos da base ou tronco cerebral. Displasia do esfenóide A displasia
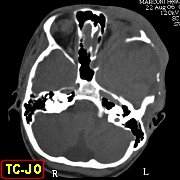
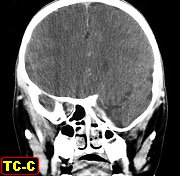
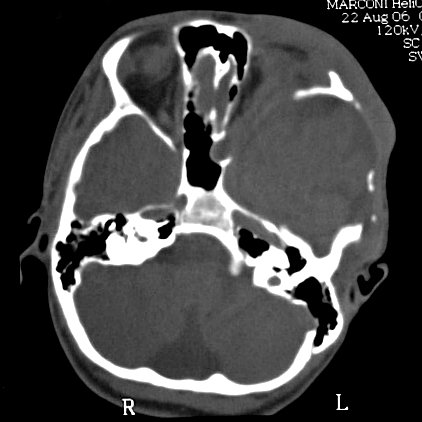
(hipoplasia, ou deficiência) da asa maior do esfenóide permite
herniação do lobo temporal no interior da órbita,
já que a grande asa faz a separação entre a parede
lateral da órbita e a extremidade anterior da fossa craniana média.
Clique para peças ósseas de base
do crânio e osso esfenóide,
e para tomografia computadorizada de
crânio normal em janela óssea.
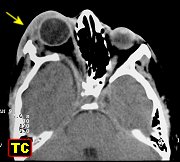
A displasia
do esfenóide é quase sempre associada a um neurofibroma plexiforme
da órbita e regiões periorbitárias. Há evidência
de que as anormalidades da órbita podem progredir, indicando que
não seriam de natureza malformativa ou displásica e sim resultariam
de erosões pelos neurofibromas. Os neurofibromas plexiformes comumente
se originam na região do ápice da órbita ou fissura
orbitária superior, na distribuição da primeira divisão
do trigêmeo. Outra localização comum é no pescoço
(25-30% dos casos). Os tumores orbitários comumente se estendem
aos seios cavernosos, nasofaringe e fissura ptérigomaxilar.
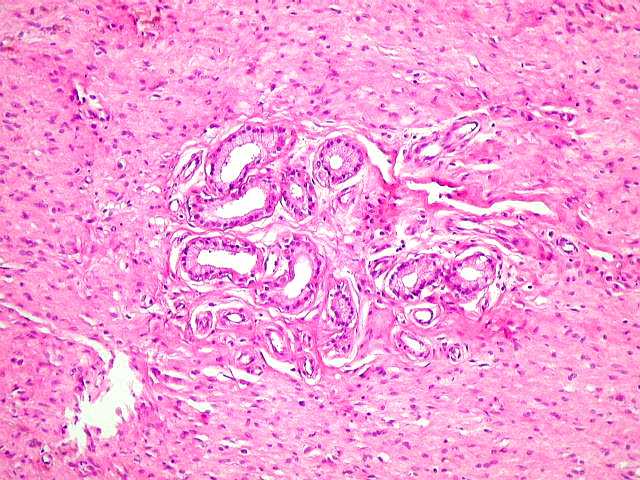
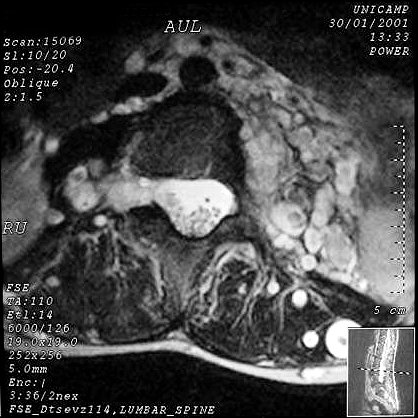
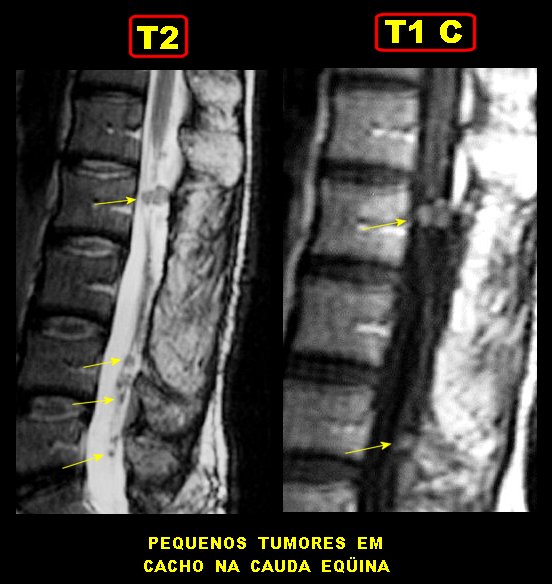
Manifestações espinais da NF1. Anormalidades de coluna ocorrem em mais de 60 % dos pacientes. Escoliose é a manifestação esquelética mais comum na NF1 (presente em cerca de 30% das crianças afetadas) e aumenta com a idade, levando a baixa estatura. Comumente resulta de displasia dos corpos vertebrais. Também se observam hipoplasia dos pedículos, apófises transversas e espinhosas, não sendo certo se as anomalias derivam de uma displasia primária do mesoderma ou de efeito dos tumores de baínhas de nervo. Alargamento de um ou mais foramens neurais é visto em mais de 60 %, geralmente pela presença de um neurofibroma naquela raiz. Neurofibromas assintomáticos intradurais extramedulares podem ocorrer em 20% dos pacientes. Tumores em ampulheta nas emergências dos nervos espinais também ocorrem em 13 a 20% dos pacientes e são histologicamente neurofibromas. Anormalidades da medula espinal são mais comuns em crianças com levoescoliose (convexidade da curvatura para a E) do que com destroescoliose. Se não é encontrada lesão intrínseca da medula, como tumor, hidrosiringomielia ou lipoma, o cone medular está no nível normal de L-2 ou acima), o filo terminal mede < 1mm no nível L5-S1, assume-se que a causa da escoliose é displasia óssea. Displasia da dura-máter e meningoceles. Meningoceles
laterais são divertículos do saco tecal, mais comuns
no nível torácico, que se projetam lateralmente através
de foramens de conjugação aumentados. As causas poderiam
ser hipoplasia dos pedículos vertebrais ou uma displasia da dura.
Uma fraqueza da dura leva a que seja focalmente esgarçada em resposta
às pulsações da pressão liquórica.
Neurofibromas
intra- ou paraespinais são outra importante anomalia na NF1. Podem
desenvolver-se em qualquer nível da coluna. São muito mais
comuns em adultos que crianças. Só 1-2% são sintomáticos,
enquanto que, na NF2, 30-40% o são. Cerca de 90% destes tumores
são extradurais, sendo metade intraforaminais. Em contraste,
na NF2, a maioria dos tumores de baínha nervosa são intradurais.
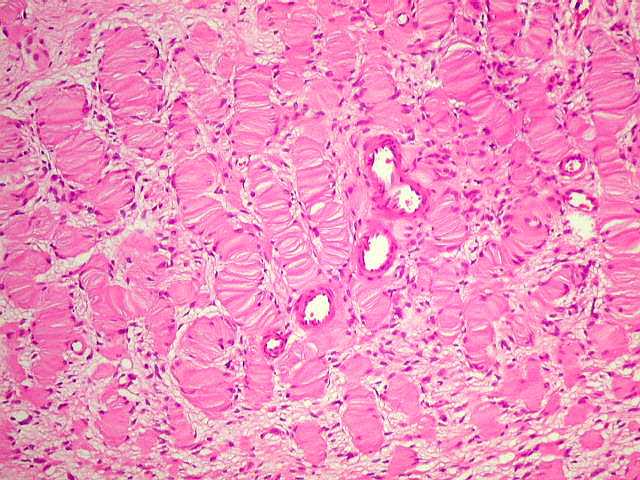
Outras lesões esqueléticas na NF1. Afinamento do córtex de ossos longos, principalmente da tíbia, causando curvaturas. Costelas em fita. Pseudoartrose. Crescimento exagerado de um dedo ou de todo um membro. Tumores fora do sistema nervoso feocromocitomas, carcinóide duodenal, rabdomiossarcoma e leucemia mielóide crônica na infância. Lesões vasculares displasia fibromuscular de grandes artérias, especialmente das artérias renais, causando hipertensão. Displasia vascular pode afetar artérias cerebrais, levando a oclusões e ao fenômeno moya-moya (circulação colateral pela região basal do cérebro). O gene NF1 está situado no braço longo do cromossomo 17 (17q12). É grande, com mais de 335 kilobases e 60 exons. A taxa de mutação, uma das mais altas na espécie humana, é estimada em 1:10.000 gametas por geração. Já foram relatadas mais de 300 mutações do gene NF1, das quais só 7% foram encontradas mais de uma vez. Não há correlações convincentes entre fenótipo e genótipo. O gene é transcrito em várias formas alternativas que são expressadas de forma diferente em neurônios e glia. A proteína resultante, chamada neurofibromina, tem duas isoformas principais, e é expressada ubiquitariamente, mas com máximas concentrações no SNC, SNP e adrenal. Observações em tumores esporádicos e associados com NF1 indicam que a neurofibromina funciona como um supressor tumoral. Em um neurofibroma, só a subpopulação de células de Schwann mostra perda de heterozigose no gene NF1, o que apóia a idéia que elas são as células progenitoras do tumor e as demais proliferam secundariamente. Relação com a mielina. O gene para a glicoproteína da mielina de oligodendrócitos está embutido no íntron 27b do gene NF1, o que se relacionaria às freqüentes anormalidades da mielina nos pacientes com NF1. A neurofibromina é necessária à mielinização normal pelas células de Schwann. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fontes:
Atlas SW. Magnetic Resonance Imaging of the Brain and Spine. 3rd Ed. Lippincott Williams & Wilkins, Philadelphia, 2002. Barkovich AJ. Pediatric Neuroimaging. 4th Ed. Lippincott Williams & Wilkins, Philadelphia, 2005. Burger PC, Scheithauer BW, Vogel FS. Surgical Pathology of the Nervous System and its Coverings. 4th Ed. 2002. Churchill Livingstone, New York. Kleihues P, Cavenee WK (eds). Tumours of the Nervous System. Pathology and Genetics. World Health Organization Classification of Tumours. IARC Press, Lyon, 2000. Osborn AG. Diagnostic Neuroradiology. Mosby, St Louis, 1994. DiPaolo DP et al. Neurofibromatosis type 1: pathologic substrate of high-signal-intensity foci in the brain. Radiology 195: 721-724, 1995. |
|
|
|
|