|
|
astrocitoma subependimário de células gigantes |
|
|
|
astrocitoma subependimário de células gigantes |
|
 |
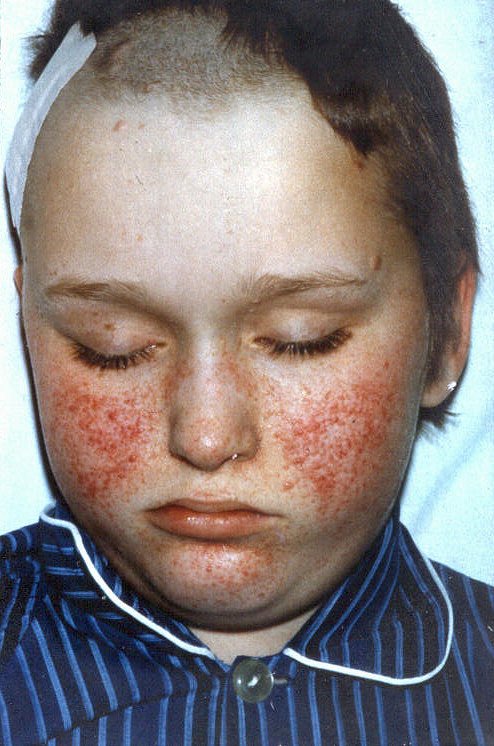 |
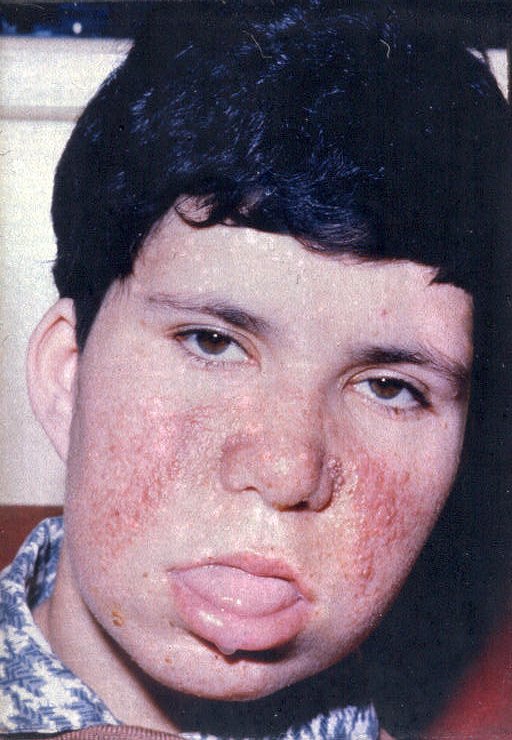
| As
lesões faciais características da esclerose tuberosa são
pápulas e nódulos numa distribuição em asa
de borboleta, conhecidas como 'adenoma sebáceo de Pringle'.
São, na realidade, minúsculos angiofibromas. Estão
presentes em mais de 90% dos pacientes acima dos 4 anos de idade e podem
também envolver o queixo e a testa.
No tronco, a lesão mais comum é a chamada 'peau chagrin' ou 'shagreen patch'. É uma placa de fibrose subepidérmica mais freqüente na região lombo-sacra. Aparece como área levemente elevada, 1-10 cm de diâmetro, com superfície comparada a pele de porco ou de elefante, ou a casca de laranja. |
|
 |
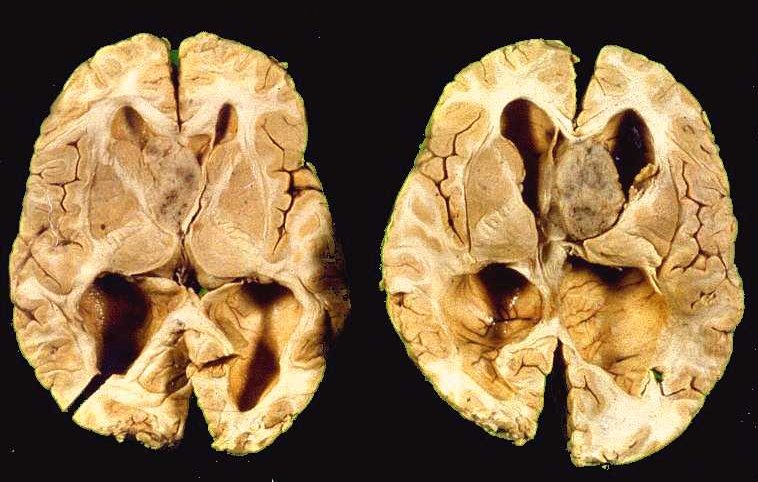 |
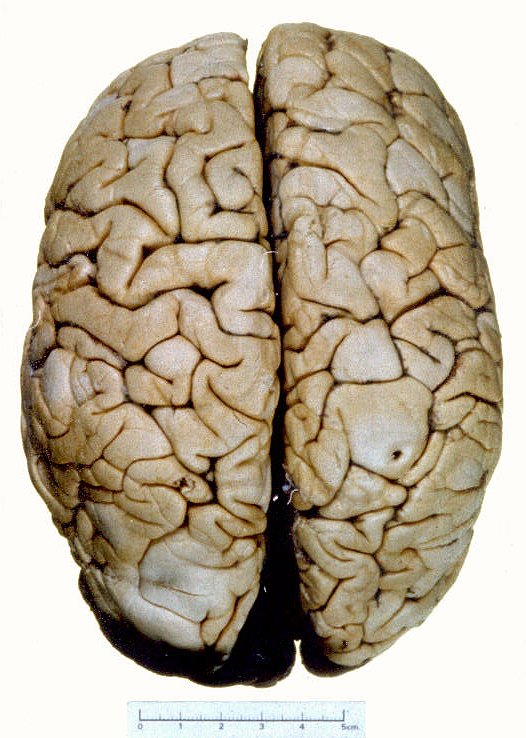
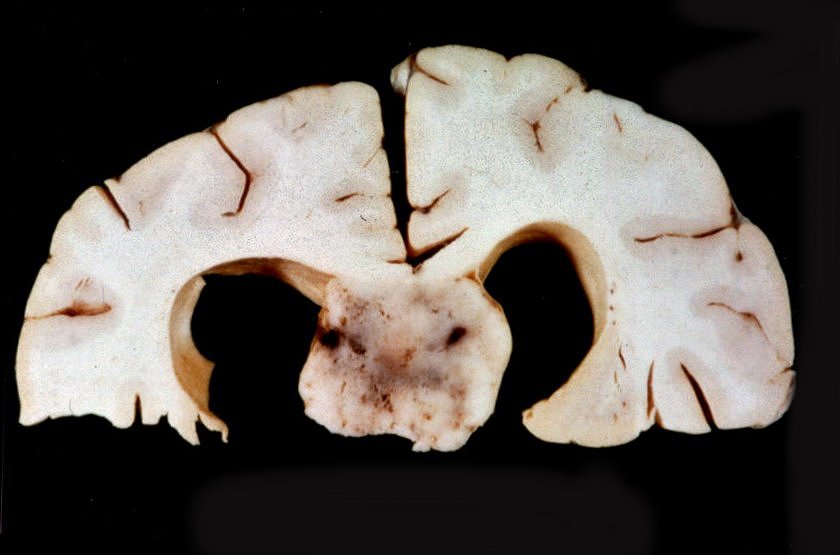
| A
E, aspecto externo da convexidade de um cérebro com esclerose
tuberosa. Notam-se áreas mais esbranquiçadas no córtex,
que correspondem aos túberes, constituídos por células
gliais ou neurônios anômalos. Alguns causam alargamento
dos giros. Na palpação a fresco, os túberes têm
consistência mais firme que áreas normais.
A D, cérebro de paciente com esclerose tuberosa cortado em plano axial (horizontal), mostrando massa tumoral bem delimitada, acinzentada, no foramen de Monro E, causando hidrocefalia bilateral. Trata-se de um astrocitoma subependimário de células gigantes. |
|
 |
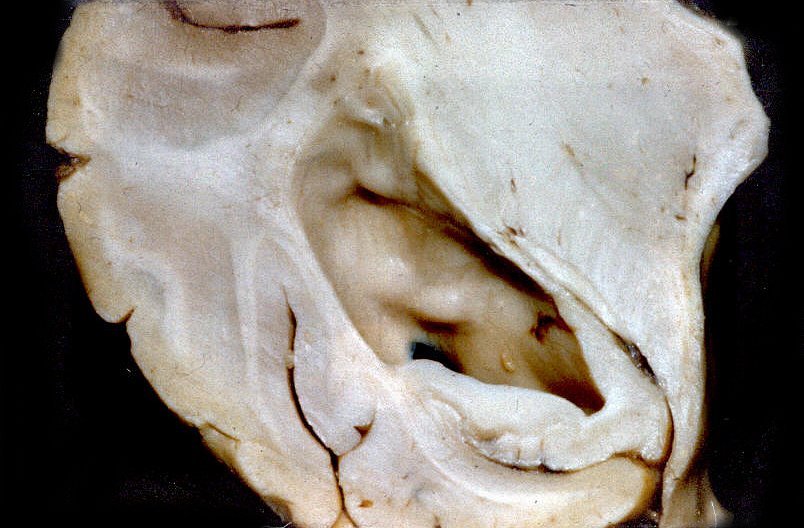 |
| A
E, astrocitoma subependimário de células gigantes
na face inferior do corpo caloso.
A D, corno inferior do ventrículo lateral, onde se notam saliências que correspondem a nódulos gliais subependimários. Estes freqüentemente calcificam. Admite-se que o astrocitoma subependimário de células gigantes origine-se em nódulos como estes. |
|
 |
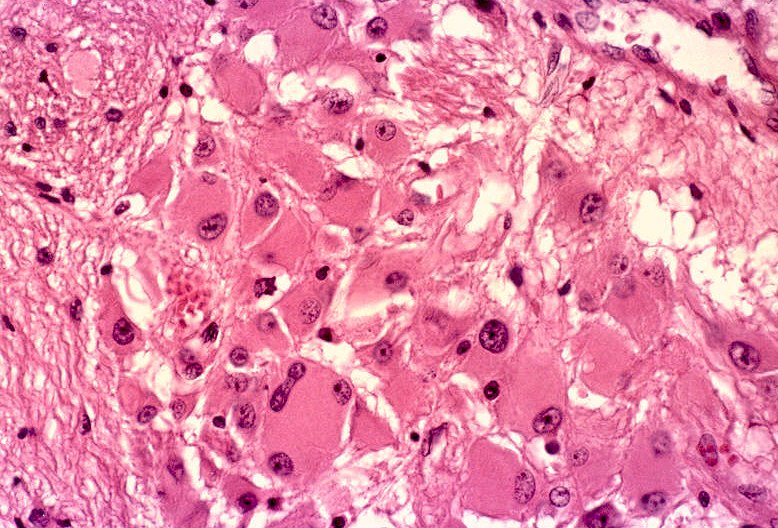 |
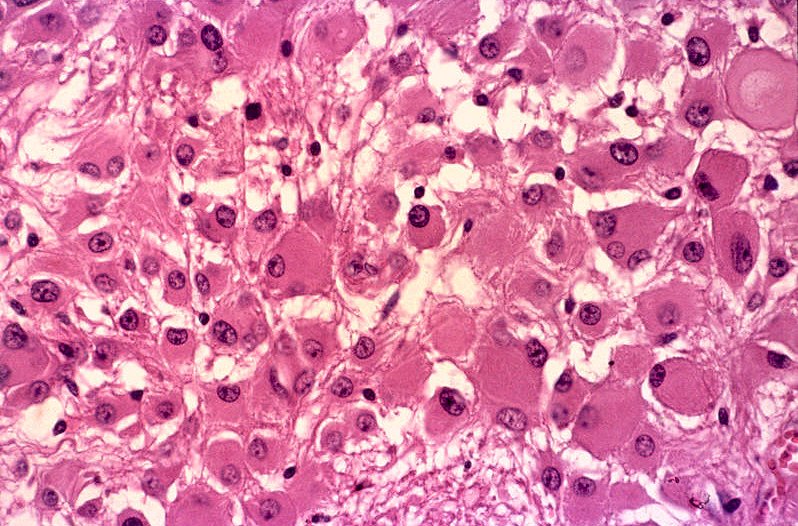
| Aspecto microscópico do astrocitoma subependimário de células gigantes. Neste exemplo, o tumor é constituído por astrócitos de aspecto gemistocítico, com citoplasma abundante e róseo e núcleo excêntrico e com poucas atipias. | |
| Esclerose
tuberosa.
O complexo
da esclerose tuberosa (doença de Bourneville, de Bourneville-Pringle,
epilóia) é um grupo de doenças autossômicas
dominantes com prevalência entre 1:5.000 e 1:10.000. Caracterizam-se
por hamartomas e neoplasias benignas que afetam o SNC: hamartomas
corticais (túberes), hamartomas glioneurais subcorticais, nódulos
gliais subependimários e o astrocitoma subependimário de
células gigantes. Manifestações extraneurais incluem
angiofibromas cutâneos (adenoma sebáceo), peau chagrin
(pele áspera como lixa), fibromas subungueais, rabdomiomas cardíacos,
pólipos intestinais, cistos viscerais, linfângio-
Principais manifestações da esclerose tuberosa. Sistema
Nervoso Central. Túberes corticais, hamartomas na substância
branca e nódulos subependimários: 90 a 100%. Astrocitoma
subependimário de células gigantes: 6 a 16%.
Clínica. Os sintomas
neurológicos mais precoces e freqüentes são epilepsia
e/ou autismo. Um certo grau de retardo mental e distúrbios de comportamento
estão quase sempre presentes, especialmente se há epilepsia.
Espasmos infantis são característicos.
Genética. História familial positiva pode ser obtida em 50% dos pacientes com esclerose tuberosa. A doença tem herança autossômica dominante com alta penetrância, mas com ampla variabilidade fenotípica. Parentes em primeiro grau de pacientes podem ter sinais clínicos ou neuroradiológicos mínimos ou formas frustas. Há
dois loci: um no cromossomo 9q34 (TSC1) e outro no cromossomo 16p13.3 (TSC2).
Várias análises demonstraram perda de heterozigose em ambos,
sugerindo que gens supressores tumorais estão envolvidos. O primeiro
gem ainda não foi isolado.
Astrocitoma Subependimário de Células Gigantes. Este tumor é parte das lesões hamartomatosas da esclerose tuberosa, e sua incidência nesta pode ser da ordem de 14%. É o tumor do SNC mais comum na ET, sendo discutível se ocorre fora dela. Nem todos tumores chegam a necessitar intervenção cirúrgica. Sintomas devidos ao tumor podem ser o primeiro sinal da esclerose tuberosa em 1/3 dos casos. Em um estudo de 34 casos de esclerose tuberosa, ASECGs confirmados por histopatologia foram clinicamente sintomáticos em 6%. Clínica. Tipicamente apresenta-se com hipertensão intracraniana, hidrocefalia e/ou hemorragia aguda nas primeiras duas décadas de vida, sem preferência por sexo. Os pacientes têm longa história de crises convulsivas devidas aos hamartomas glioneurais corticais (túberes) e heterotopias na substância branca. A localização mais comum é parede do ventrículo lateral próximo ao foramen de Monro. Pequenos nódulos hamartomatosos subependimários no sulco caudo-talâmico coexistem com o tumor. Macroscopia. Os tumores mostram nódulos, cistos e calcificações, e a consistência é firme devida ao estroma glial. Microscopicamente, o tumor é OMS grau I, e mostra população celular heterogênea numa matriz astroglial fibrilar: células pequenas fusiformes, células poligonais ou gemistocíticas e células globóides, que podem lembrar neurônios. Pode haver células multinucleadas. Os núcleos são pleomórficos e podem ter nucléolo proeminente. Os vasos podem ser dilatados, com paredes finas ou hialinizadas, e sangram espontaneamente. Apesar da morbidade operatória, estes tumores têm prognóstico favorável, mesmo com atipias, mitoses, necrose e proliferação vascular (rara). Raramente recidivam, e mesmo nestes não se relatou transformação maligna. Imunohistoquímica.
GFAP e proteína S-100 podem ser positivas em proporção
variável das células. Algumas proteínas de citoesqueleto
associadas a neurônios podem ser demonstradas, p. ex. neurofilamentos
e tubulina. Proteínas gliais e neuronais podem ser identificadas
em populações celulares semelhantes. Parece haver coexistência
de células gliais (astrócitos e epêndima), neurônios
(uma fração menor de células) e diferenciação
neuronal-glial mista. Algumas células semelhantes a neurônios
mostram em microscopia eletrônica grânulos secretórios
e estruturas que lembram sinapses.
Túberes corticais podem ser detectados por TC ou RM e associam-se a epilepsia. Microscopicamente, consistem de células gigantes (como as vistas nos ASECGs) e neurônios dismórficos, estendendo-se da meninge à substância branca, com perda da laminação cortical, gliose e calcificação de vasos. O córtex em volta tem arquitetura normal. Na substância branca adjacente há perda da mielina, agrupamentos heterotópicos de neurônios anormais e células gigantes. Os neurônios displásicos mostram perda da orientação radial, da forma piramidal, arborização dendrítica aberrante, espinhas dendríticas em densidade reduzida, projeções axonais diminuídas e acúmulo de fibrilas no pericário. As células gigantes vistas nos túberes têm heterogeneidade semelhante à vista nos ASECGs. Imunohistoquimicamente células gigantes nos túberes podem ter fenótipo glial, neuronal ou misto. Muitas expressam nestina, um filamento intermediário encontrado em células neuro-epiteliais precursoras, gliais e neuronais. Algumas expressam GFAP; outras, com morfologia idêntica, não. Vários marcadores neuronais são também encontrados. Formação de sinapses pode ser observada, mas não é consistente. Principal fonte: O.D. Wiestler et al. Tuberous sclerosis complex and subependymal giant cell astrocytoma. in Tumours of the Nervous System. Pathology and Genetics. Kleihues P, Cavenee WK, eds. IARC Press, Lyon, 2000. |
| Mais sobre
esclerose tuberosa e ASECGs em:
Casos de neuroimagem; Caso neuropatológico |
| Módulo Neuro - Página Inicial | Outros módulos | e-mails : gradanat@fcm.unicamp.br___gradanat@unicamp.br |