|
|
Pineocitoma, Pineoblastoma. Texto didático ilustrado |
|
|
|
Pineocitoma, Pineoblastoma. Texto didático ilustrado |
|
| Nesta página
: tumores do parênquima da pineal: macroscopia;
pineocitoma : exames de imagem, histologia, imunohistoquímica, microscopia eletrônica; pineoblastoma : exames de imagem, histologia, imunohistoquímica. Clique para tumores germinativos do SNC; Pineal normal, histologia, imunohistoquímica. Obs. as figuras estão linkadas às paginas originais dos respectivos casos. Para mais detalhes e imagens, clique nas figuras. |
| Os tumores
do parênquima da pineal
formam um contínuo desde os pineocitomas considerados benignos ou
grau I da OMS, até os pineoblastomas que são altamente agressivos
(grau IV). Entre eles estão os tumores de diferenciação
intermediária (graus II ou III).
Historicamente, o termo pinealoma foi cunhado por Krabbe em 1923 e abrangia vários tipos de tumores na topografia da pineal, incluindo os germinomas. Em 1947, Dorothy Russell e Nathan Friedman separaram os germinomas dos tumores do parênquima da pineal propriamente dito. Incidência. Tumores da região pineal são coletivamente raros (< 1% dos tumores do sistema nervoso central nos países ocidentais). A distribuição por tipos é cerca de 35% para tumores germinativos, 28% para tumores do parênquima pineal, e 28% para gliomas, incluindo ependimomas. Na Ásia, a incidência dos tumores da região pineal é maior, chegando a cerca de 3,2% no Japão. Naquela parte do mundo, tumores germinativos respondem por 80% do total, sendo a metade constituída por germinomas. Os tumores do parênquima pineal correspondem a 12% e gliomas a 6,5%. Idade. Adultos tendem a apresentar tumores benignos (pineocitomas, idades entre 36 e 47 anos). Crianças são afetadas pelas variedades mais agressivas. Pineoblastomas têm idades médias entre 12 e 18 anos. Clínica. O quadro inicial mais comum é síndrome de hipertensão intracraniana por obstrução do aqueduto. Embaçamento visual pode decorrer do edema de papila. Compressão dos colículos superiores pode manifestar-se como paralisia do olhar conjugado para cima (síndrome de Parinaud). Pode haver ptose palpebral ou diplopia, por lesão do núcleo do oculomotor, e nistagmo por compressão do fascículo longitudinal medial. Para esquema da neuroanatomia, clique. Imagem. Os métodos atuais não permitem distinção segura entre os tumores do parênquima pineal entre si ou de outros, como os tumores germinativos. O diagnóstico diferencial entre eles é histopatológico. Os pineocitomas caracteristicamente são arredondados, bem demarcados, mais compressivos que infiltrativos. Os pineoblastomas tendem a ser mais infiltrativos, e podem mostrar disseminação meníngea. (Contudo, há considerável superposição). Pode haver calcificações na TC. Na RM as lesões dão isossinal à substância cinzenta em todas as seqüências e se impregnam por contraste. Histopatologia. Pineocitomas são bem diferenciados, moderadamente celulares, compostos de células lembrando pineocitos. As células neoplásicas são uniformes, com núcleos ovalados, arranjadas difusamente ou em padrão lobular. As rosetas pineocitomatosas são uma feição característica, e consistem de grandes áreas fibrilares arredondadas circundadas por células neoplásicas. Com técnicas argênticas, ou imunohistoquímica para neurofilamento, as células podem mostrar prolongamentos orientados para o centro da roseta e terminados em bulbos. Vasos são delicados, sem proliferação endotelial, e não há necrose. Mitoses são muito raras ou ausentes. Pineoblastomas são tumores pouco diferenciados de células pequenas, redondas, com núcleos densos e escuros, e citoplasma escasso de limites imprecisos (entram na categoria de tumores de pequenas células azuis ou small blue cell tumors). A relação núcleo-citoplasma é alta, como para outros tumores malignos. São, portanto, indistinguíveis morfologicamente dos meduloblastomas e outros tumores neuroectodérmicos primitivos (PNETs) do sistema nervoso central. Podem ocorrer pseudorrosetas de Homer Wright, menores que as rosetas pineocitomatosas. Há variável atividade mitótica e áreas de necrose. Os vasos são geralmente de paredes finas, mas pode haver proliferação endotelial. Comumente, há invasão da glândula pineal e das meninges. Pode haver diferenciação para fotorreceptores, na forma de rosetas verdadeiras ou de Flexner-Wintersteiner (com lúmen). Tumores de diferenciação intermediária ainda não têm critérios bem definidos para designação como grau II ou III. As feições que parecem correlacionar-se com o comportamento biológico dos tumores do parênquima pineal são a taxa de proliferação (avaliada pela contagem de mitoses ou positividade para Ki-67), e o grau de diferenciação neuronal, demonstrada por estudos ultraestruturais ou imunohistoquímicos. O pineocitoma e pineoblastoma representam os extremos deste espectro. Imunohistoquímica. Diferenciação neuronal nos tumores do parênquima pineal é avaliada por sinaptofisina (SNF), enolase neurônio-específica (NSE), e proteína de neurofilamento (NF). O marcador neuroendócrino cromogranina A também pode ser usado. A positividade é forte e citoplasmática nos pineocitomas. Nos pineoblastomas a marcação é variável, menos intensa e pode ser negativa para alguns marcadores. Tratamento. Em pineocitomas, o principal objetivo é a ressecção completa do tumor por via infratentorial supracerebelar ou occipital transtentorial. Para pineoblastomas, a abordagem baseia-se em cirurgia, radioterapia e quimioterapia. A ressecção completa ainda é o melhor indicativo de bom prognóstico, com trabalhos mostrando sobrevida de 10 anos de 15% se restou tumor significativo, contra 100% para ausência de tumor residual. Irradiação cranioespinal (visando controlar disseminação por via liquórica) reduz o risco de recidivas locais e distantes. Radioterapia em crianças pequenas deve ser retardada para minorar déficits neurocognitivos. Quimioterapia deve ser usada como adjuvante, pois não é de alta eficiência isoladamente. Tumores de grau intermediário devem ser ressecados tão completamente quanto possível, pois tumor residual pode evoluir para grau mais alto. O prognóstico depende do tipo histológico, estadiamento quando do diagnóstico e resposta ao tratamento inicial. Pineocitomas têm prognóstico favorável com sobrevida de 5 anos entre 86 e 91%. Para tumores de grau intermediário isto varia entre 49 a 62%. Para pineoblastomas, em que é comum disseminação liquórica, fica em 10%, portanto pior que para meduloblastomas (atualmente na casa dos 60%). Fonte:
Vasiljevic A, Fèvre-Montange M, Jouvet A. Pineal Parenchymal
Tumors. In Perry A, Brat DJ (eds). Practical Surgical Neuropathology.
Churchill Livingstone Elsevier, 2010. pp 151-63.
|
|
|
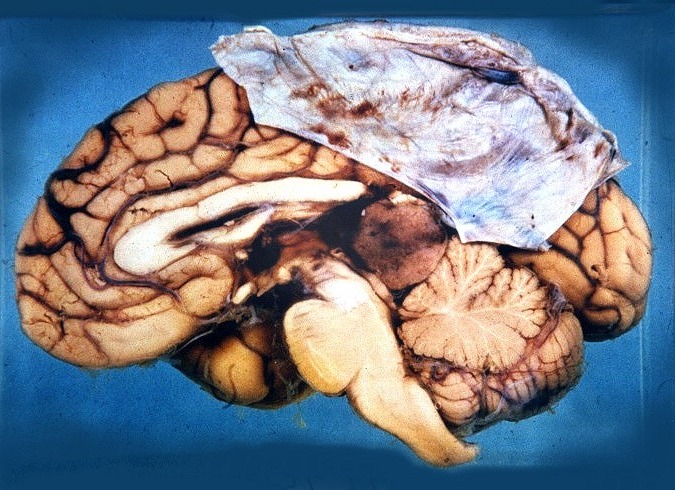
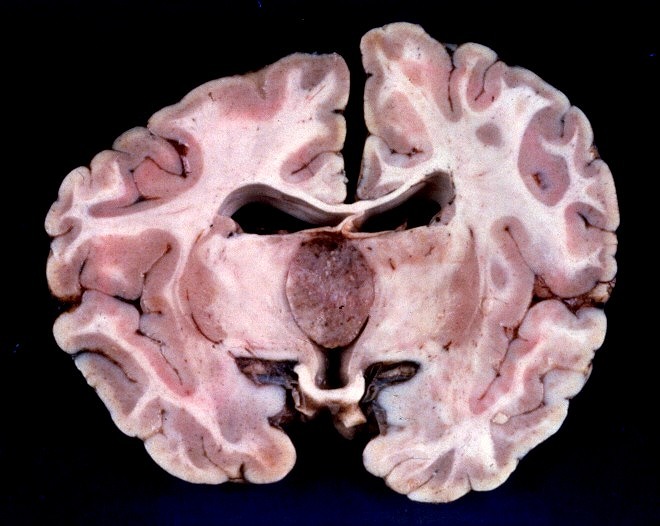
| Slides escaneados. Atualmente é muito difícil, senão impossível, obter peças de autópsia como as abaixo. Estas imagens provêm de antigas coleções de slides que datam de mais de 30 anos. Foram escaneadas e aqui reproduzidas para proveito geral e, principalmente, para que não se percam. No espécime da esquerda, em corte sagital, o tipo de tumor não está especificado, mas qualquer das principais neoplasias da pineal (germinomas, pineocitomas ou pineoblastomas) seriam candidatas prováveis. No espécime à direita há menção de que se tratava de um pineoblastoma. Para mais detalhes e mais imagens, clique nas fotos. | |
 |
 |
|
|
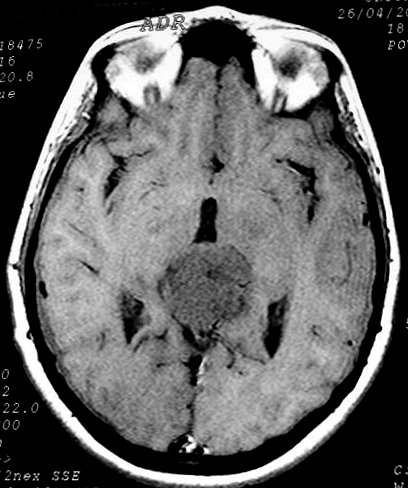
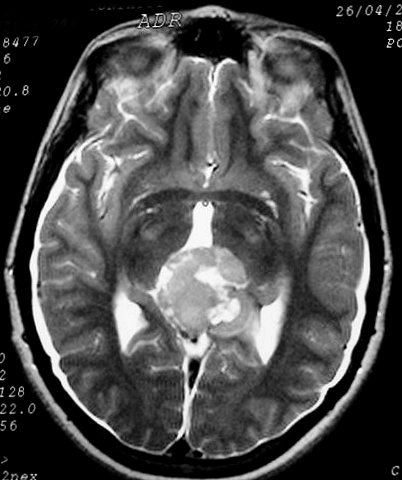
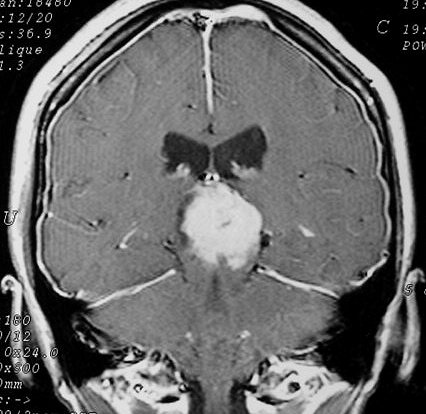
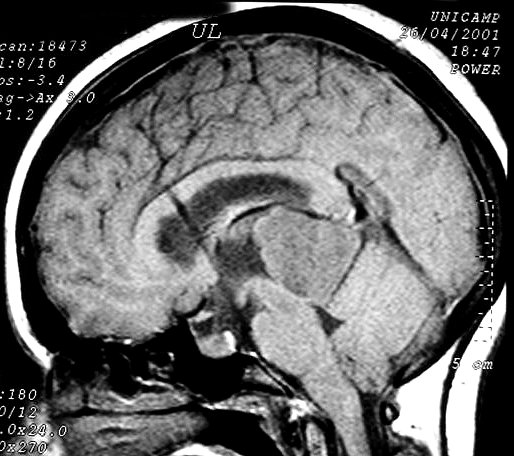
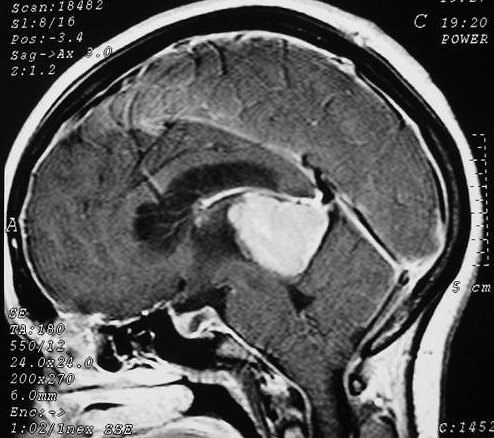
| Pineocitoma. F. 24 a. Tumor sólido bem delimitado na topografia da glândula pineal. Na TC, isodenso ao parênquima cerebral, com forte impregnação por contraste. Na RM, hipointenso em T1, hiperintenso em T2, captante. No corte sagital, substitui a pineal, eleva o esplênio do corpo caloso, deprime a placa quadrigêmea e comprime o aqueduto cerebral. | |||
| TC axial, sem contraste | TC com contraste | RM axial, T1 sem contraste | T2 |
 |
 |
 |
 |
| Coronal, T1 com contraste | Sagital, T1 sem contraste | T1 com contraste |
 |
 |
 |
| Para exame histológico deste caso, clique. | ||
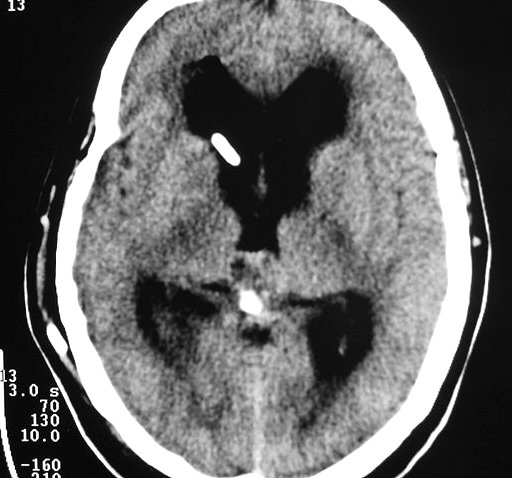
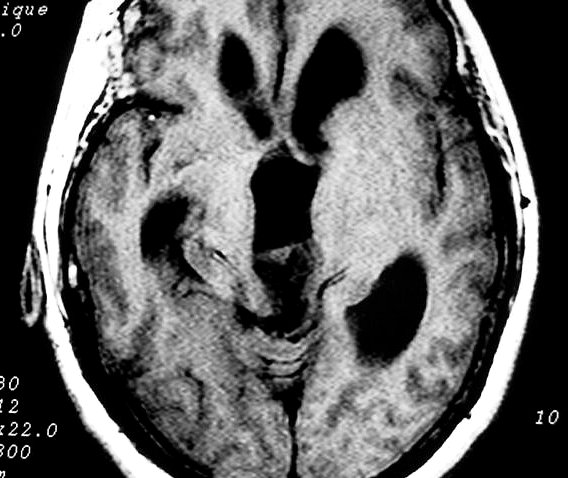
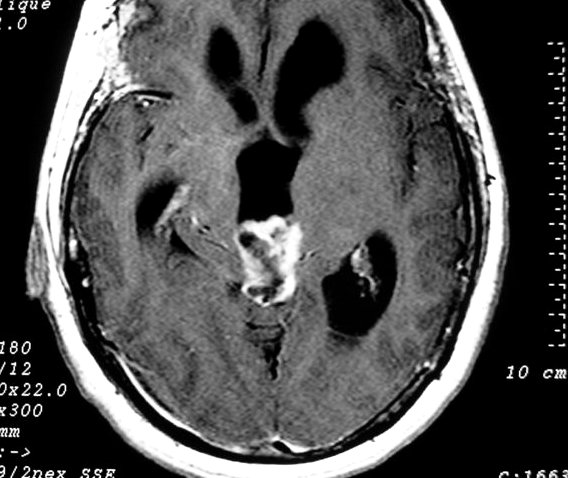
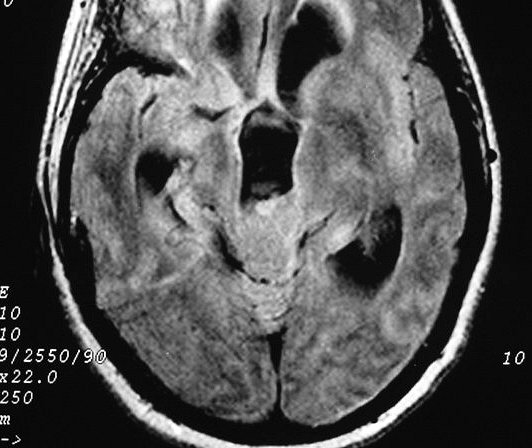
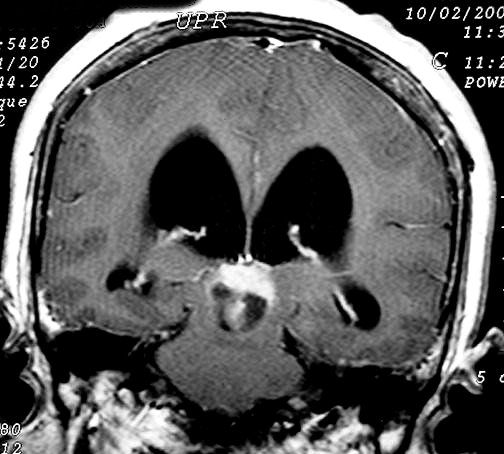
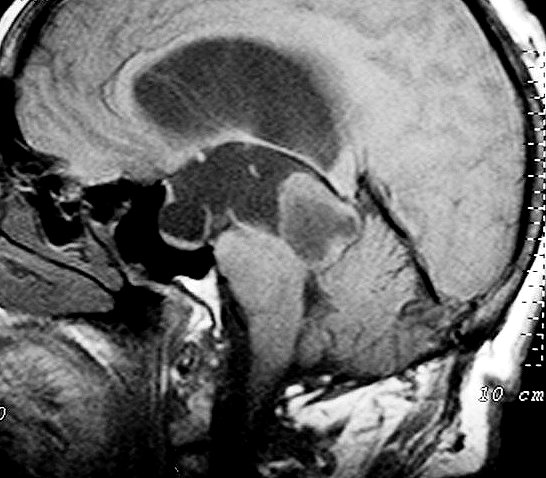
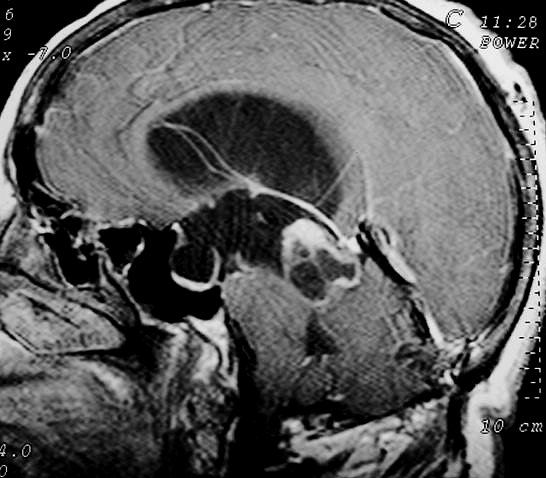
| Pineocitoma. F. 56 a. Tumor na topografia da pineal, com cistos envolvidos por componente sólido que se impregna. Na TC sem contraste, a área calcificada deve corresponder à pineal original, não demonstrável na RM. A real extensão do tumor só pode ser avaliada com contraste. No FLAIR axial, observa-se hipersinal no conteúdo dos cistos, indicando alta concentração de proteínas. Nos cortes sagitais, tumor comprime o aqueduto de Sylvius e causa grande hidrocefalia. A distensão do III ventrículo leva a insinuação do mesmo no interior da sela túrcica, com compressão da hipófise. | |||
| TC sem contraste | RM axial, T1 sem contraste | T1 com contraste | FLAIR |
 |
 |
 |
 |
| Coronal, T1 com contraste | Sagital, T1 sem contraste | T1 com contraste |
 |
 |
 |
| Para exame histológico deste caso, imunohistoquímica e microscopia eletrônica, clique. | ||
|
|
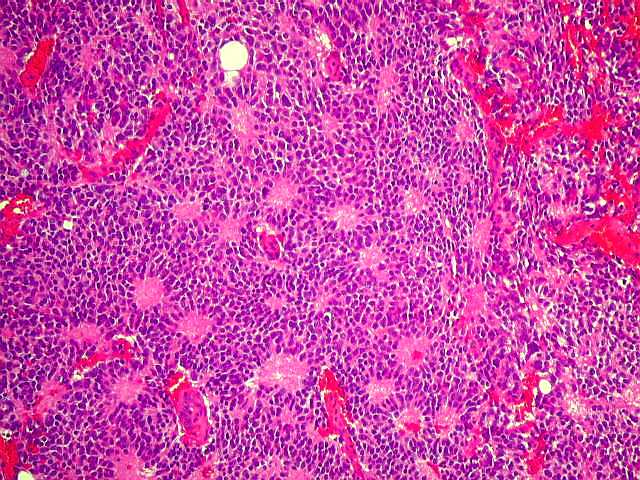
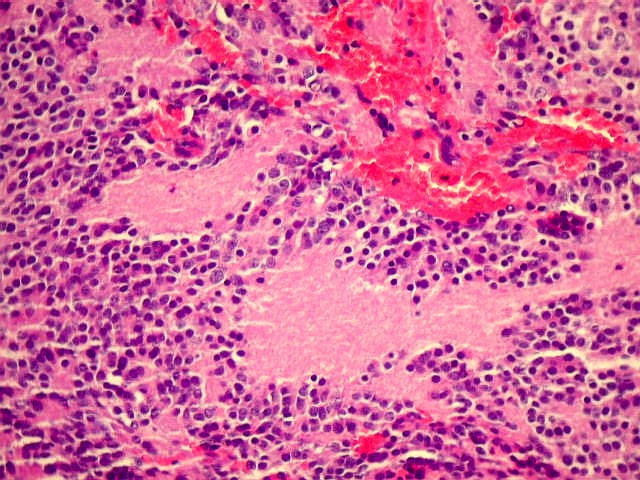
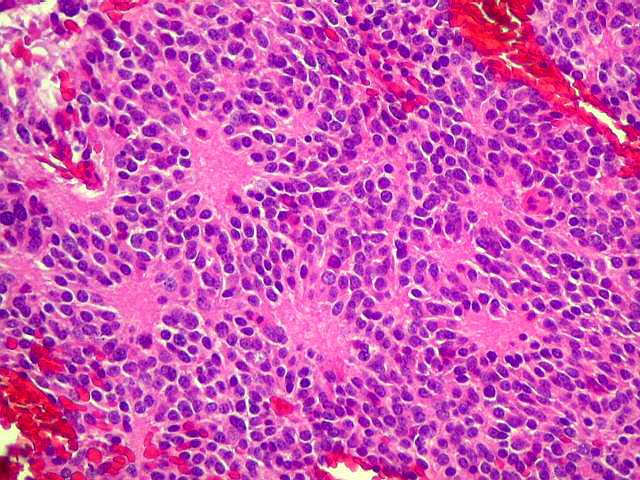
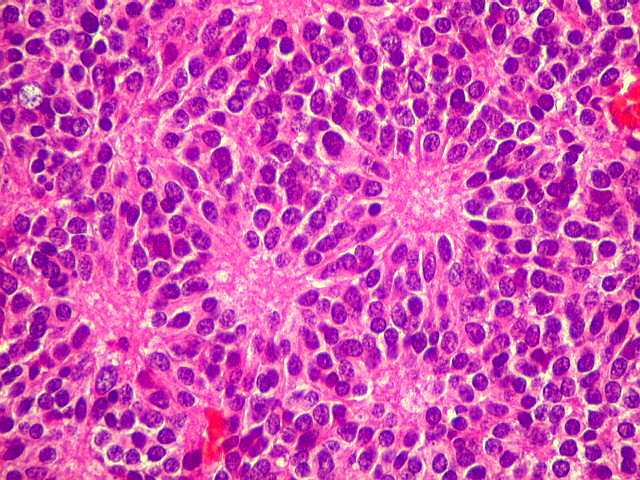
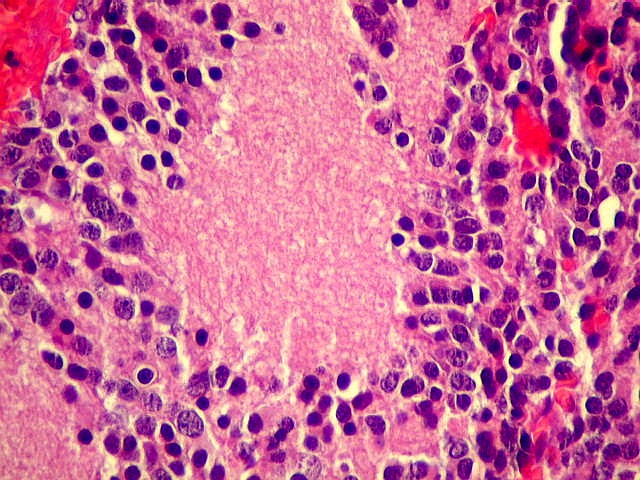
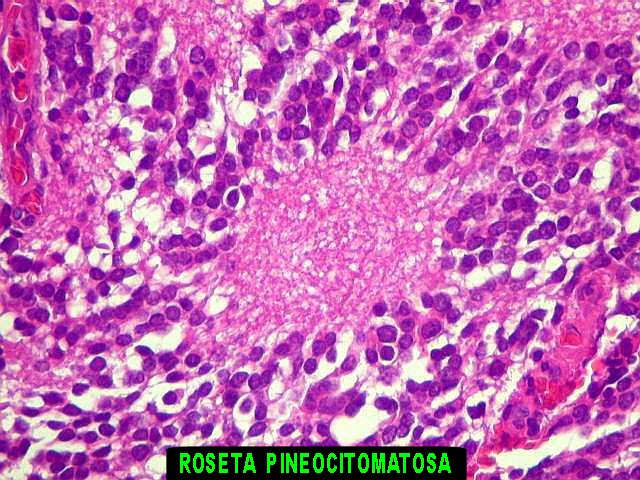
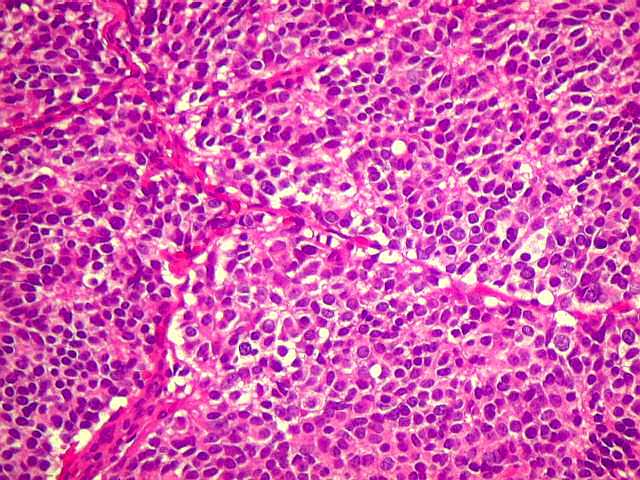
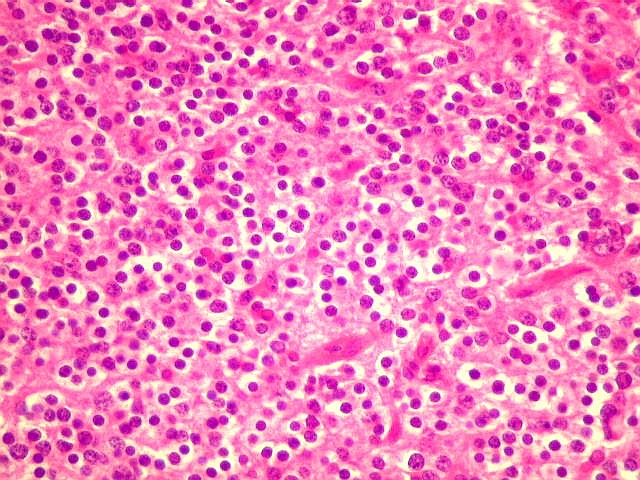
| Pineocitoma HE - Rosetas pineocitomatosas. Principal elemento diagnóstico deste raro tumor de pineocitos, as rosetas ditas 'pineocitomatosas' lembram as rosetas de Homer Wright, mas são maiores, com ampla área central anucleada composta por prolongamentos das células neoplásicas. Em volta, as células têm núcleos regulares, com cromatina densa, mas geralmente sem atipias. Mitoses e necrose não são observadas. | |
 |
 |
 |
 |
 |
 |
 |
 |
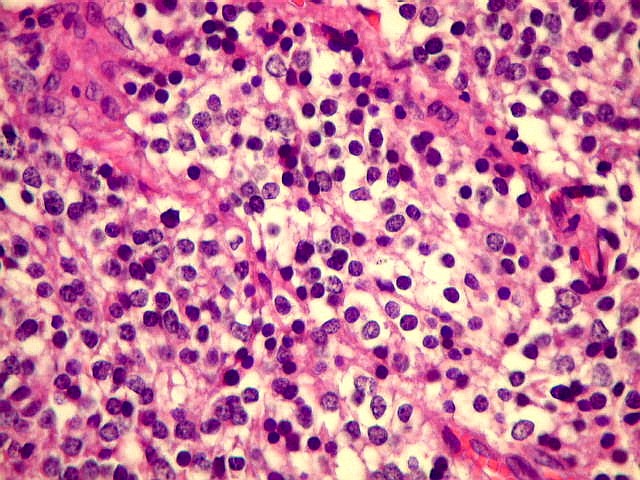
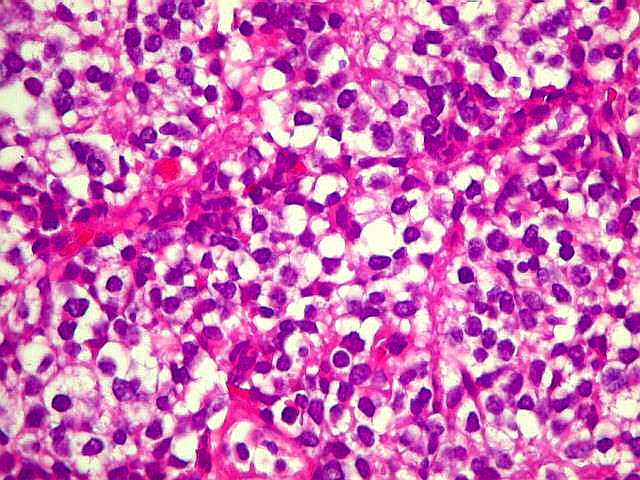
| Áreas sólidas podem lembrar oligodendroglioma. Em áreas do tumor que não mostram rosetas, as células neoplásicas dispõem-se compactamente entre delicados vasos, que podem lembrar a arquitetura 'em tela de galinheiro' dos oligodendrogliomas. Por vezes, vacuolização do citoplasma pode simular o clássico aspecto 'em ovo frito' dos oligodendrócitos, normais ou neoplásicos. | |
 |
 |
 |
 |
 |
| Concreções calcáreas. Podem ser encontradas, aumentando a semelhança com oligodendrogliomas. | Atipias nucleares. De modo geral são raras nos pineocitomas. |
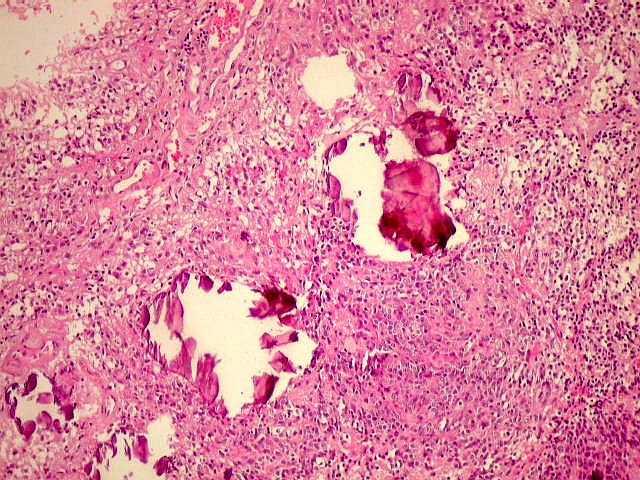 |
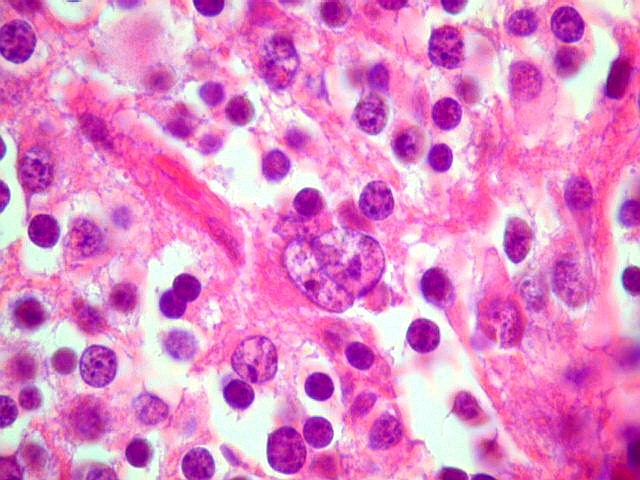 |
| Esfregaço com pseudorrosetas. Encontro de formações rosetóides em esfregaços do tumor pode antecipar o diagnóstico de pineocitoma. | |
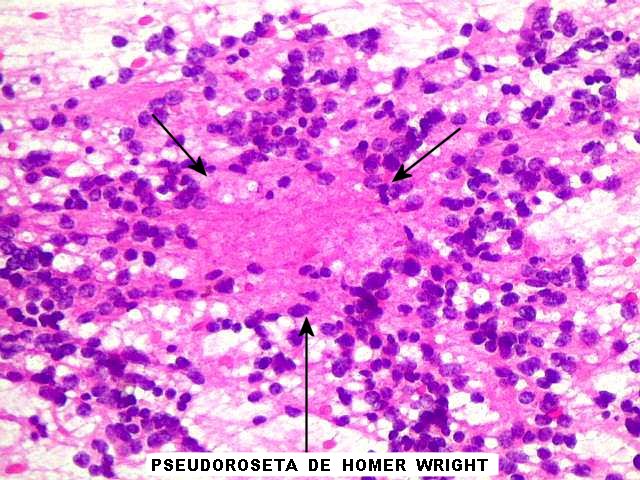 |
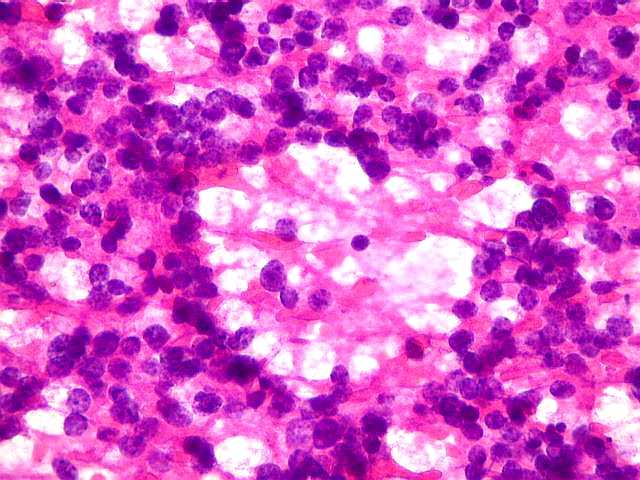 |
|
|
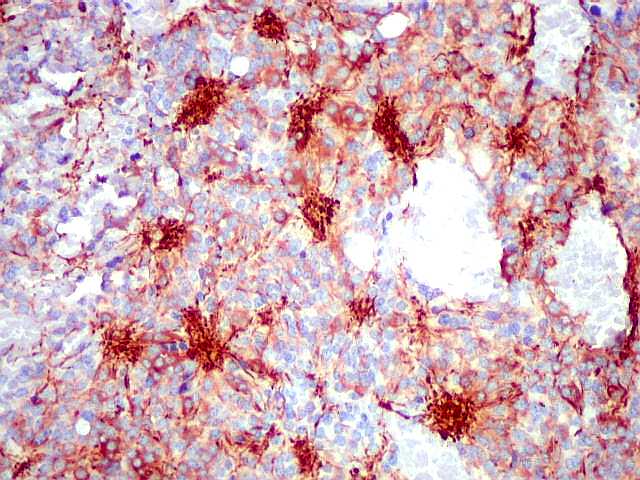
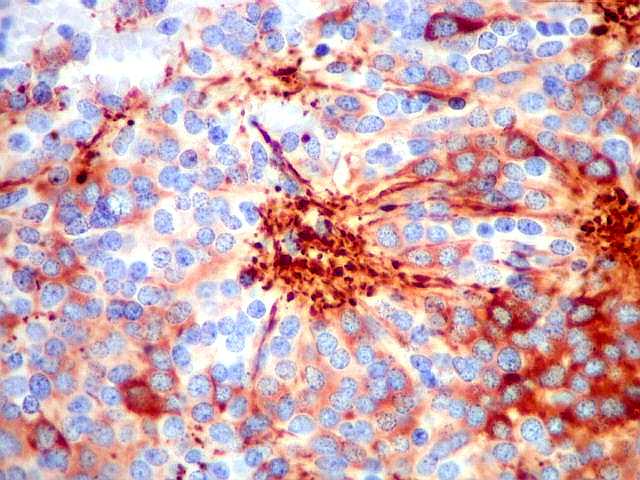
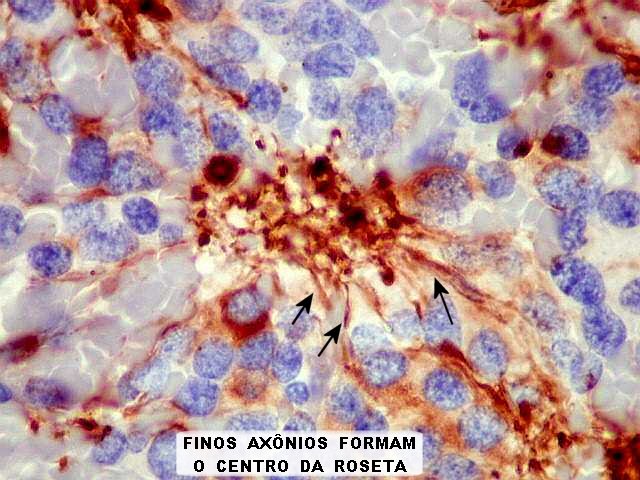
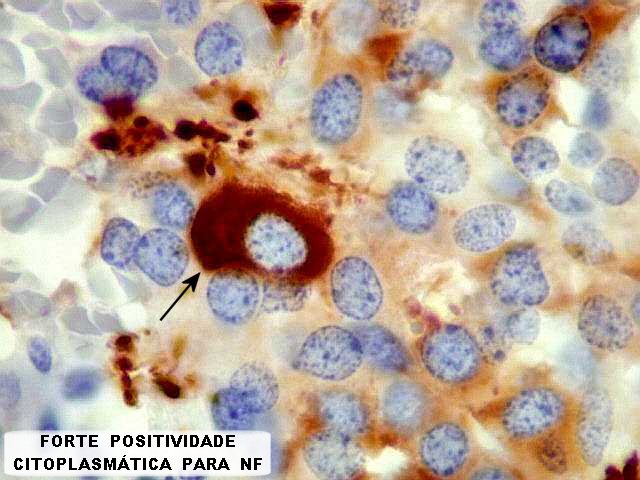
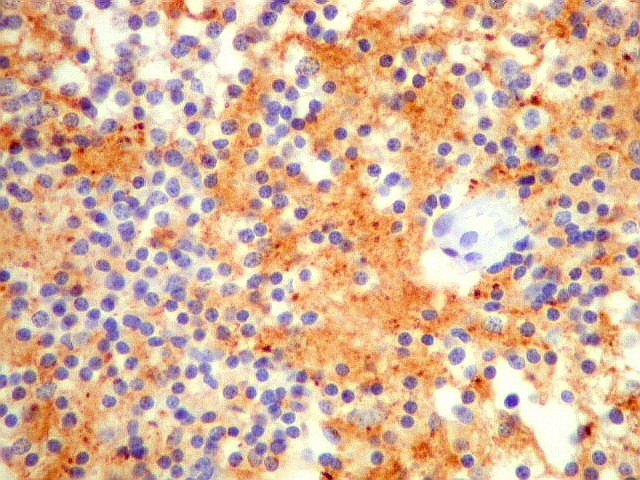
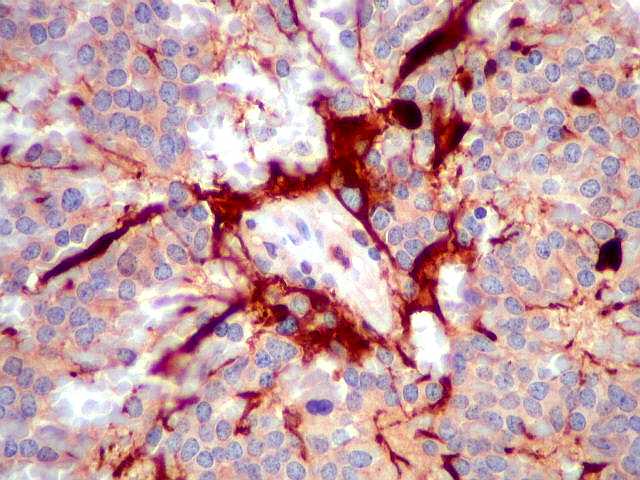
| NF. Proteína de neurofilamento, um marcador de linhagem neuronal, é positiva principalmente no centro das rosetas pineocitomatosas, por vezes permitindo visualizar os finos prolongamentos celulares. Em alguns casos, é possível demonstrar as expansões bulbosas, também presentes em pineocitos normais. A marcação pode ocorrer no corpo celular, contrastando com a negatividade em células vizinhas. | ||
 |
 |
 |
 |
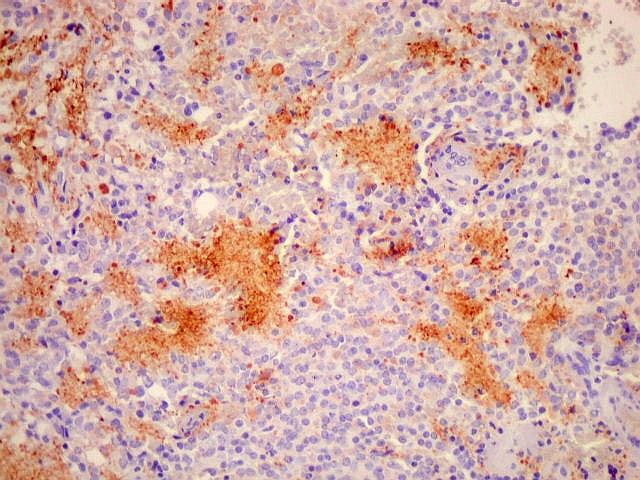 |
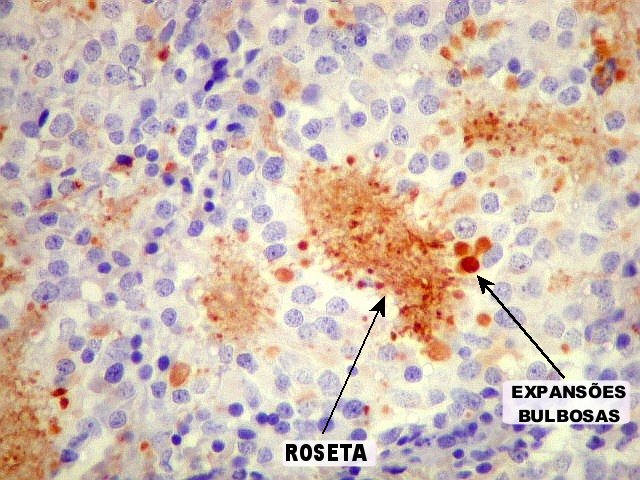 |
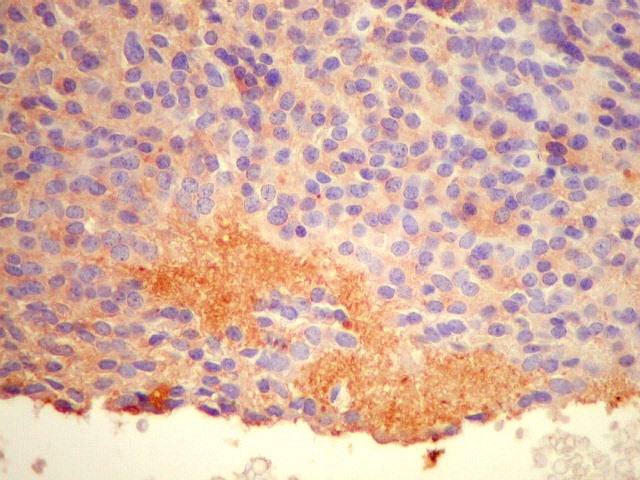
| SNF. Sinaptofisina, outro marcador de linhagem neuronal ou neuroendócrina, repete os resultados com NF. A positividade é mais forte no centro das rosetas, mas também é observada no citoplasma e no interstício entre as células. | |
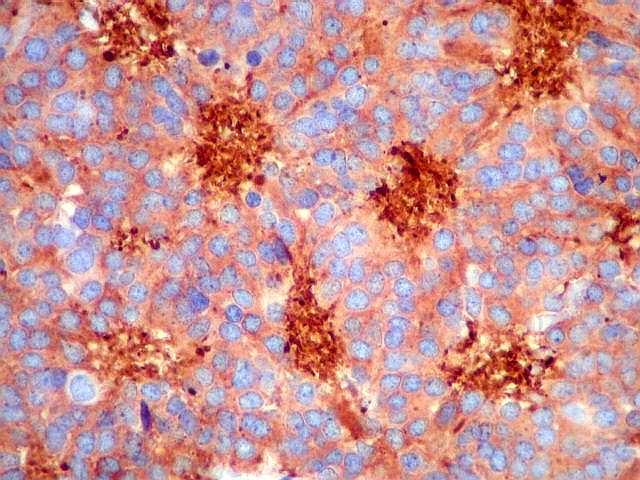 |
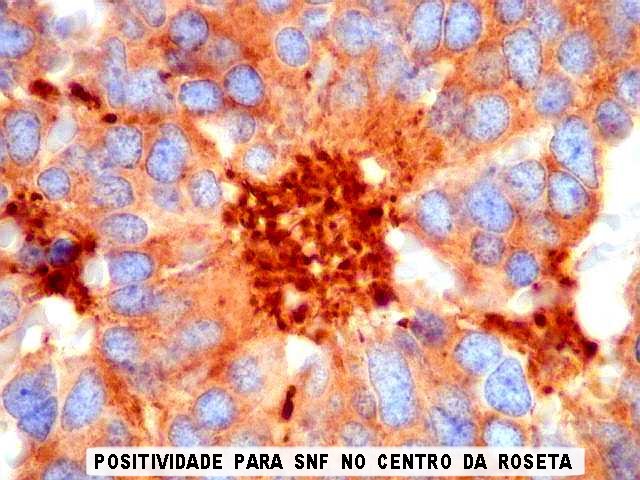 |
 |
 |
 |
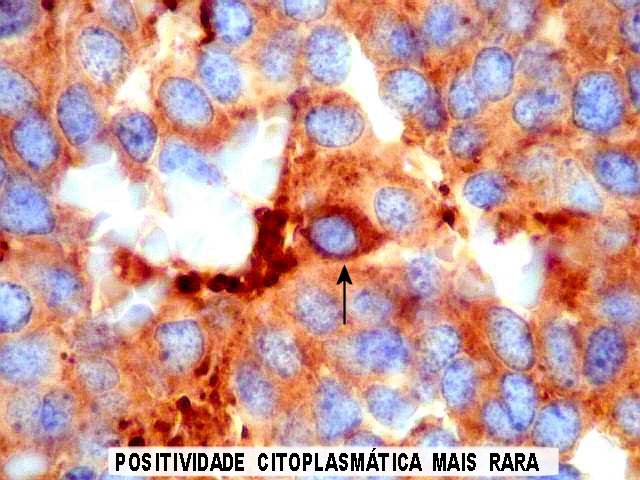
| Cromogranina. Resultado semelhante aos acima, mas menos forte ou constante. | ||
 |
 |
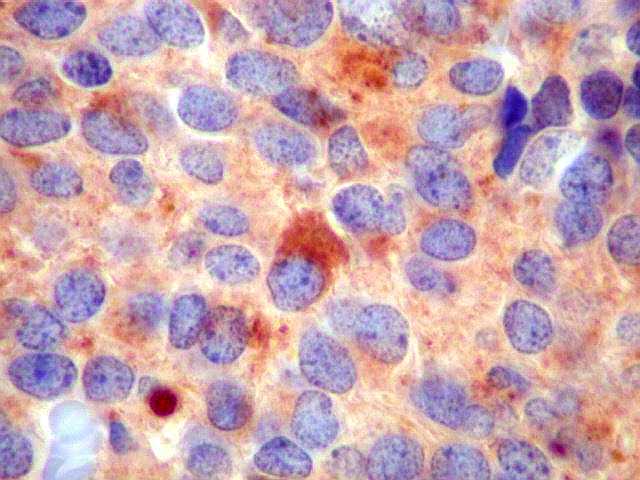 |
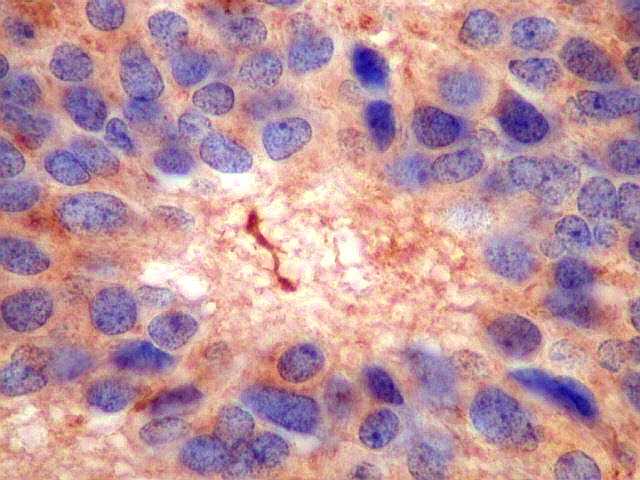
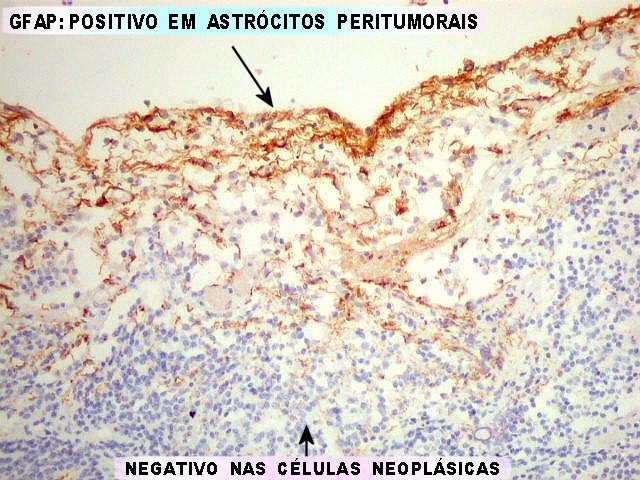
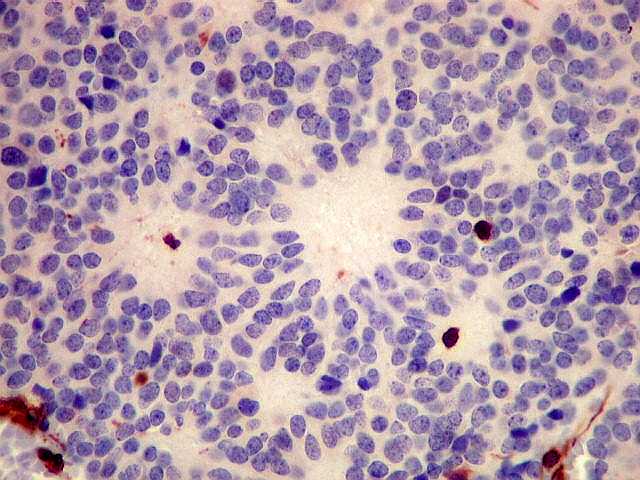
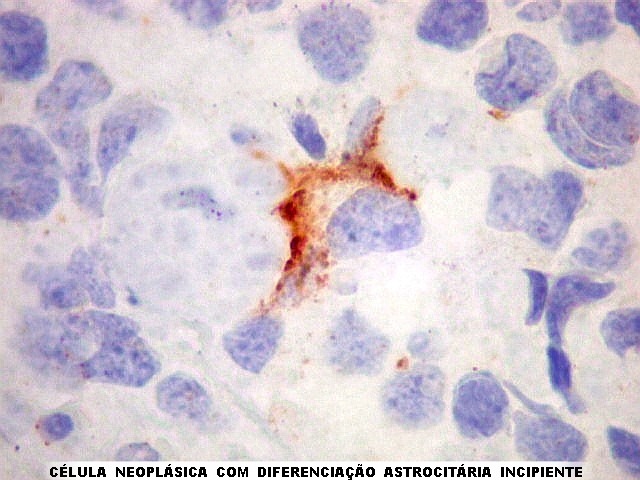
| GFAP. Este marcador de astrócitos é negativo no tumor. Pode ser observada marcação de astrócitos peritumorais ou em tecido infiltrado pelo tumor. Ocasionalmente, pode ser vista marcação em células com morfologia compatível com a de elementos neoplásicos, pela semelhança dos núcleos e escassez de citoplasma, sem prolongamentos. Admite-se que estas células possam exibir diferenciação divergente em sentido glial, como visto no neurocitoma central (1) (2), no meduloblastoma e em outros PNETs (clique para exemplos). | ||
 |
 |
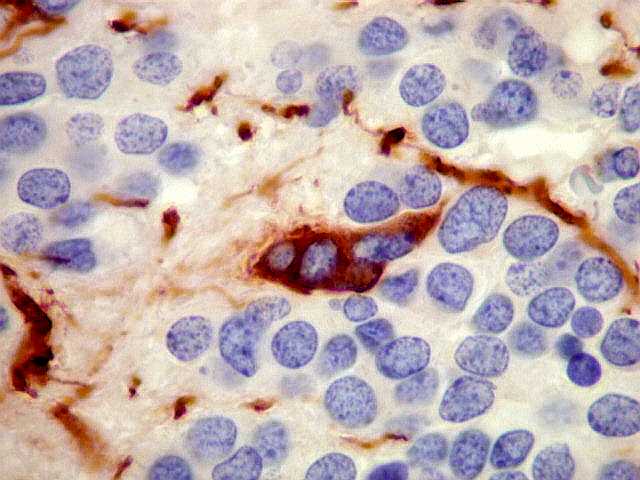 |
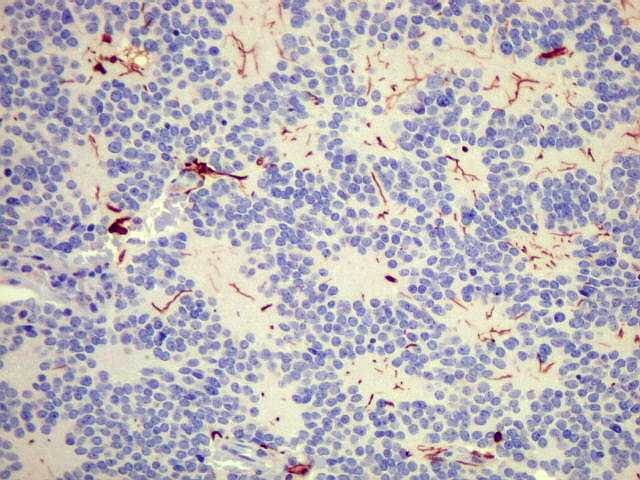
| VIM. Positiva só em vasos, negativa nas células tumorais. | ||
 |
 |
 |
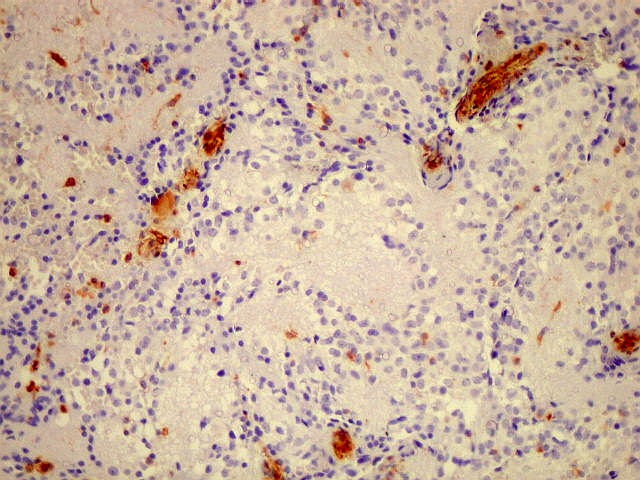
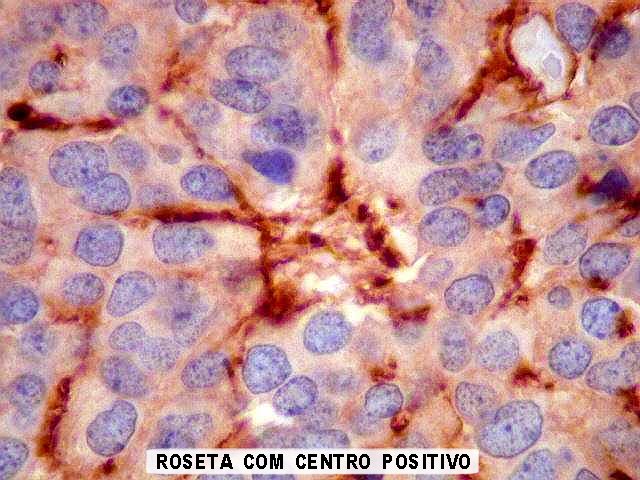
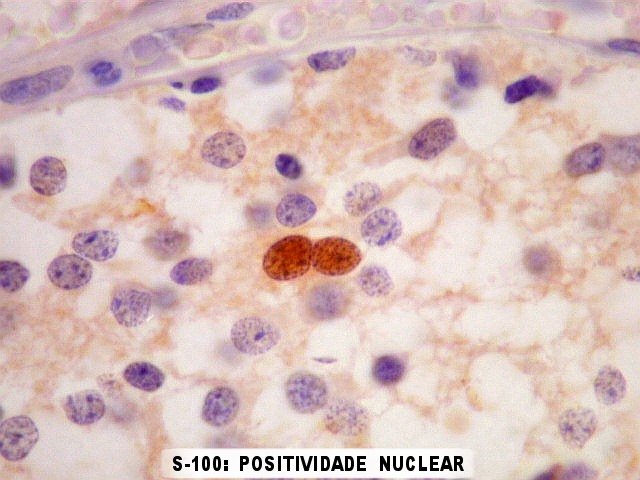
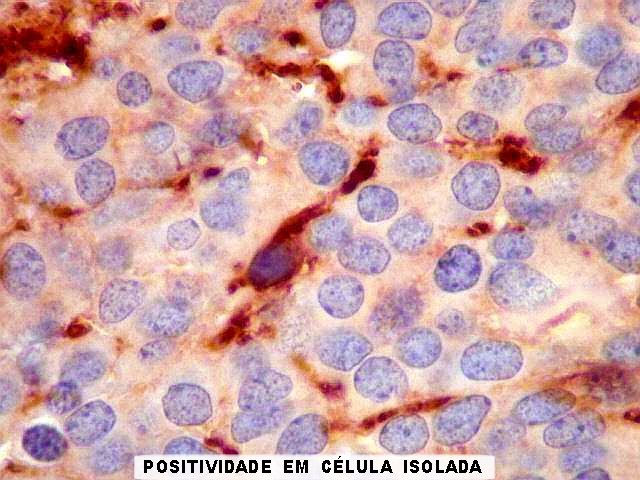
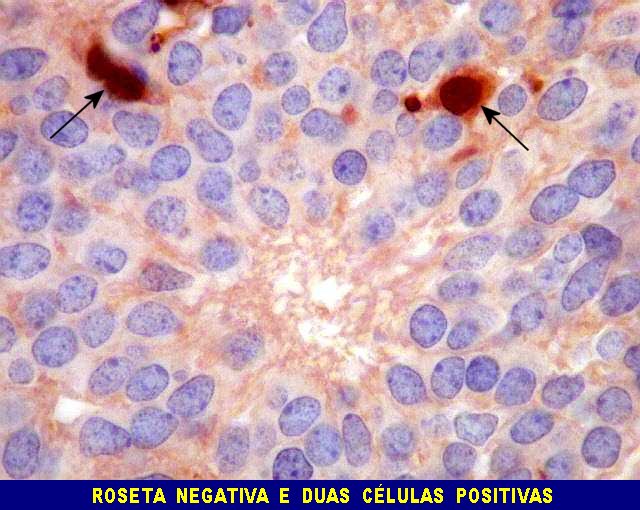
| S-100. De modo geral negativo nas células tumorais, com algumas células positivas. Podem representar elementos com diferenciação glial, como já comentado para GFAP, acima. | |
 |
 |
 |
 |
 |
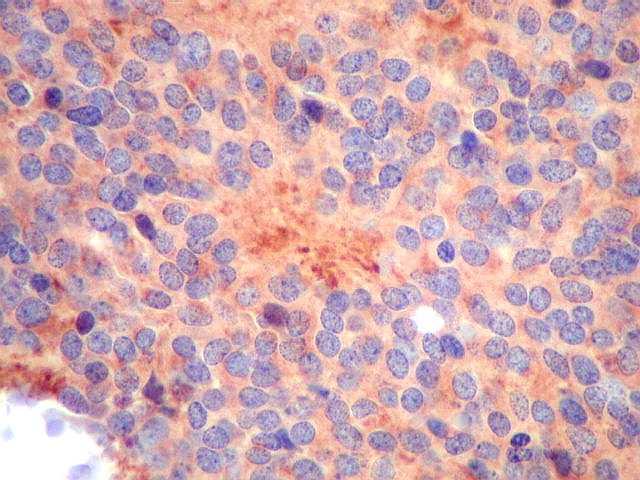
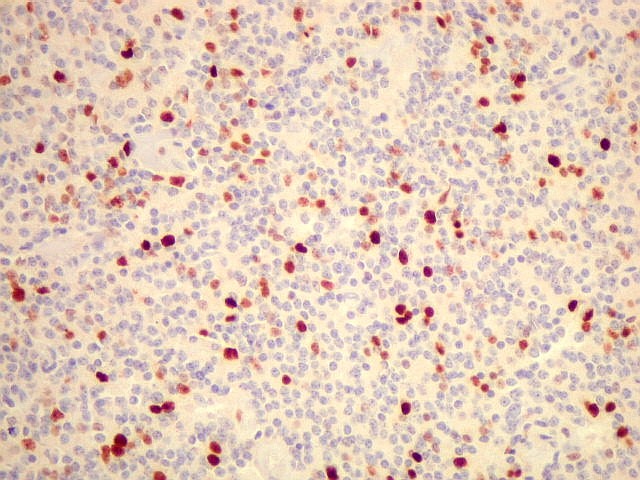
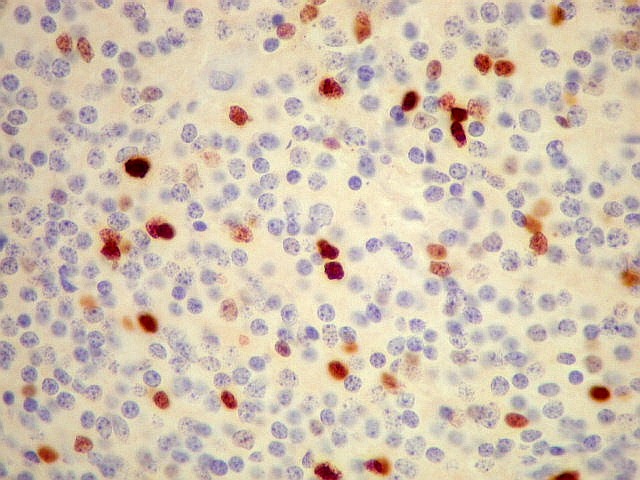
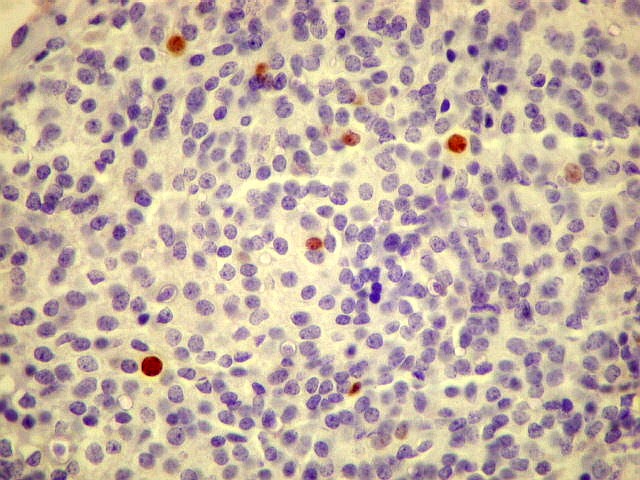
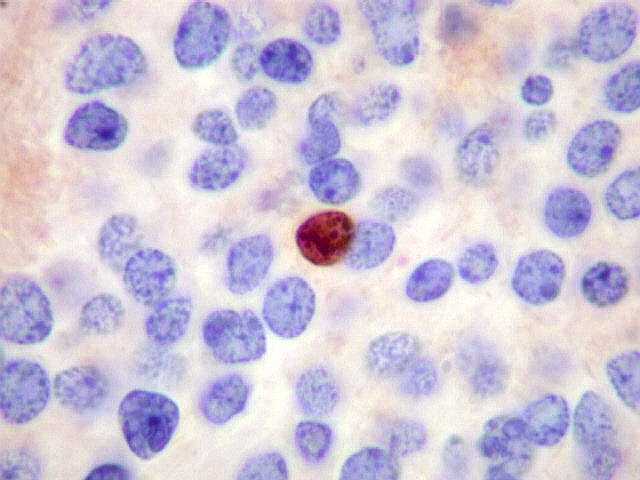
| Ki-67. Positividade variou entre os casos, desde cerca de 1% a 10%. Na literatura descrevem-se valores baixos, consistentes com o grau I atribuído a este tumor pela OMS. Valores mais altos podem indicar formas com diferenciação intermediária e maior agressividade. | ||
 |
 |
 |
 |
 |
p53. Totalmente negativo na única amostra testada. |
 |
||
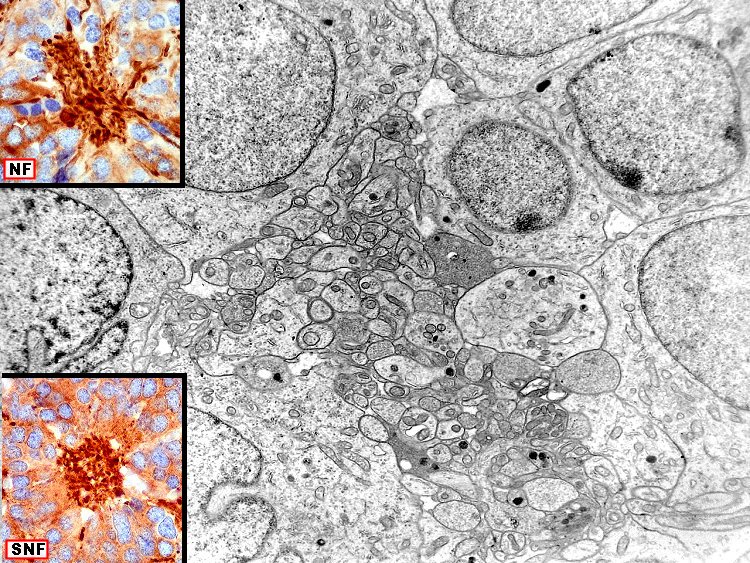
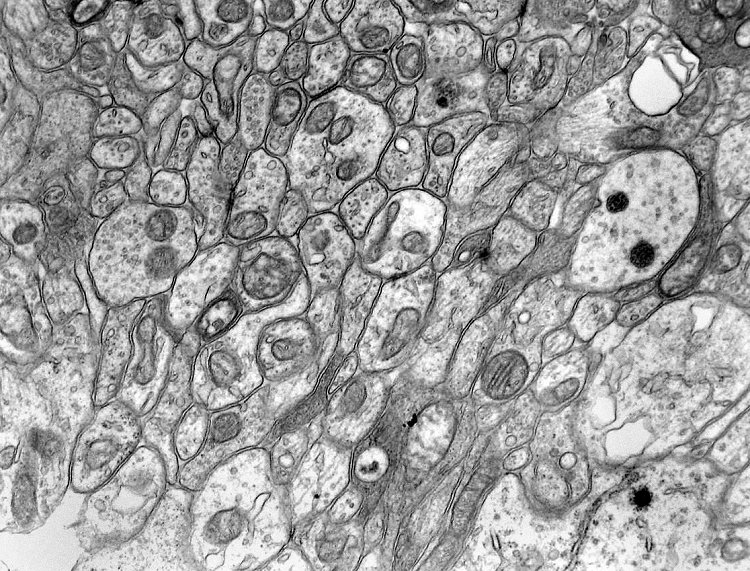
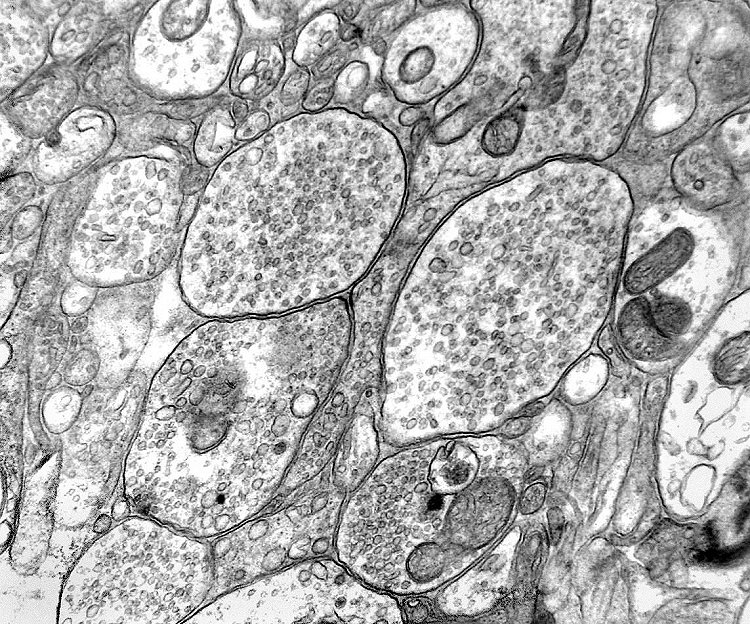
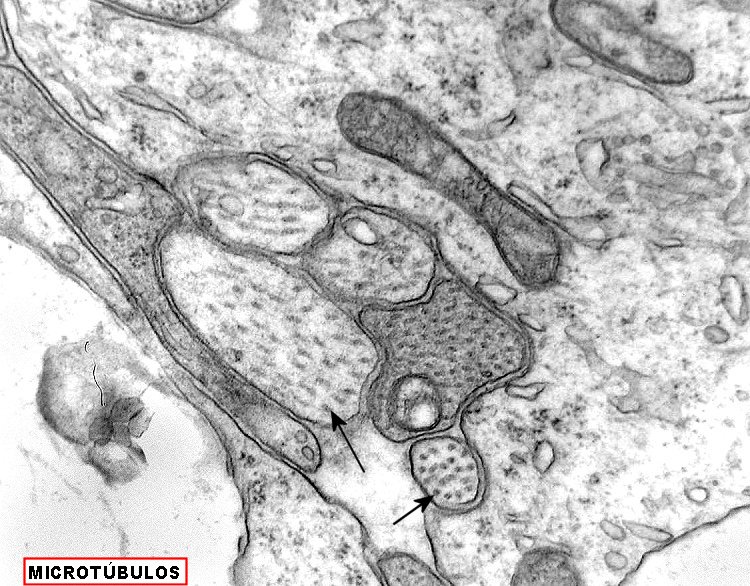
| Pineocitoma - Microscopia eletrônica. Realizada em um só caso, corrobora a natureza neuronal do tumor. O centro das rosetas tem estrutura semelhante a neurópilo, com perfis de prolongamentos citoplasmáticos intimamente apostos, vários contendo vesículas do tipo sináptico e microtúbulos. | |
 |
 |
 |
 |
|
|
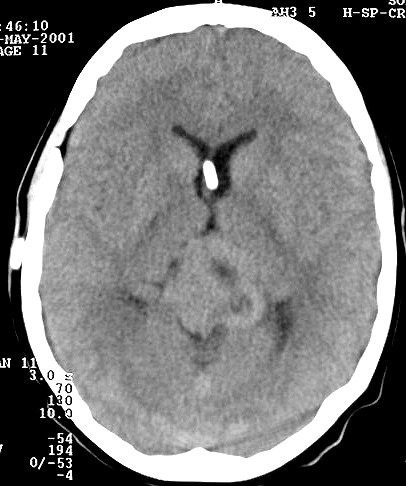
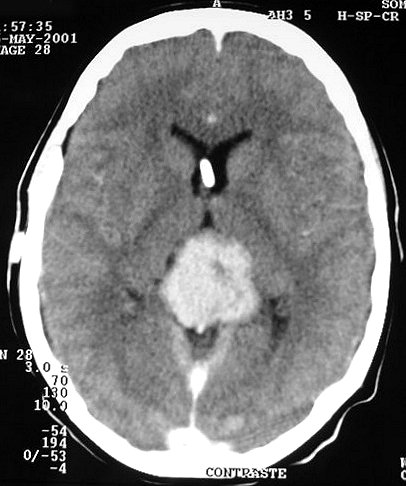
| Pineoblastoma em Tomografia Computadorizada. F. 20 a. Tumor em 3 datas, antes de intervenção cirúrgica que estabeleceu o diagnóstico de pineoblastoma. Crescimento lento do tumor, mantendo características de boa delimitação. É espontaneamente hiperdenso em relação ao cérebro. | ||
| 1996, sem contraste | 1998, sem contraste | 2002, com contraste |
 |
 |
 |
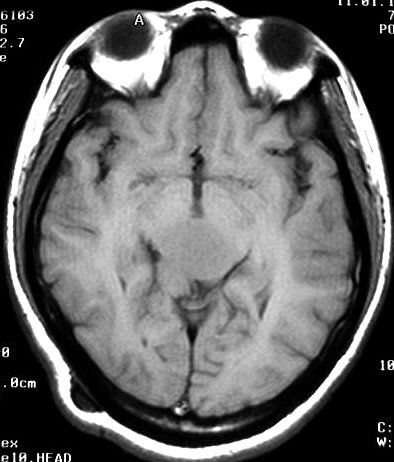
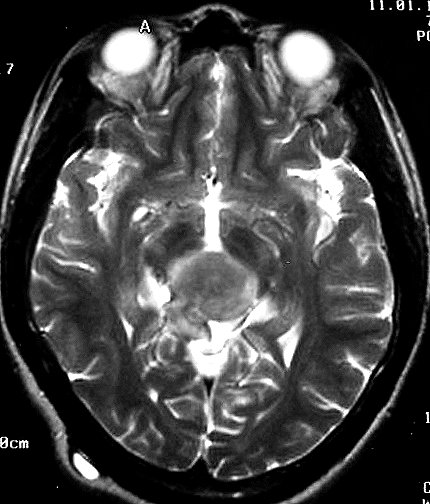
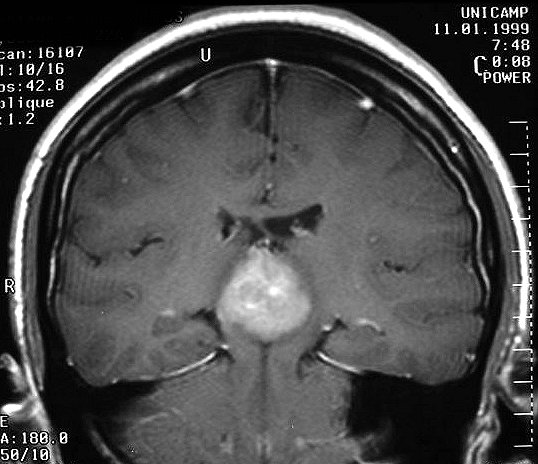
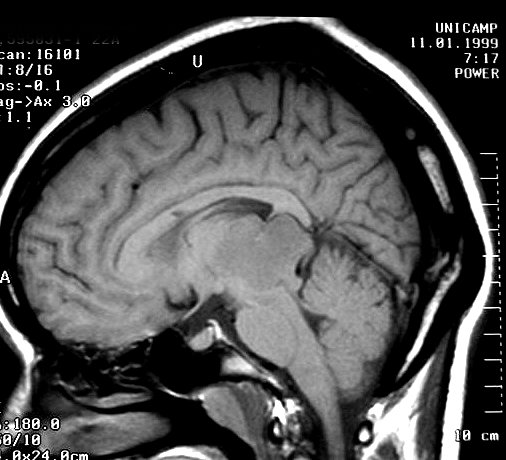
| Pineoblastoma em Ressonância Magnética. Mesmo caso, em exame de 1999. A neoplasia mostra crescimento predominantemente expansivo, mas há perda da nitidez dos limites, tendendo a infiltrar o tecido nervoso, especialmente o tegmento mesencefálico, como observado no sagital em T1 com contraste. | ||
| T1 SEM CONTRASTE | T1 COM CONTRASTE | T2 |
 |
 |
 |
| CORONAL, T1 contraste | SAGITAL, T1 | T1 contraste |
 |
 |
 |
| Para exame histológico deste caso, clique (link). | ||
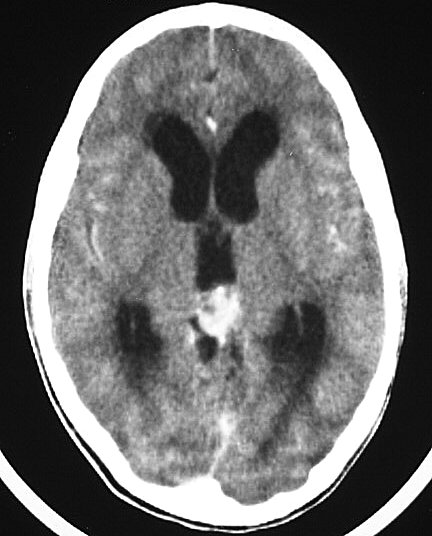
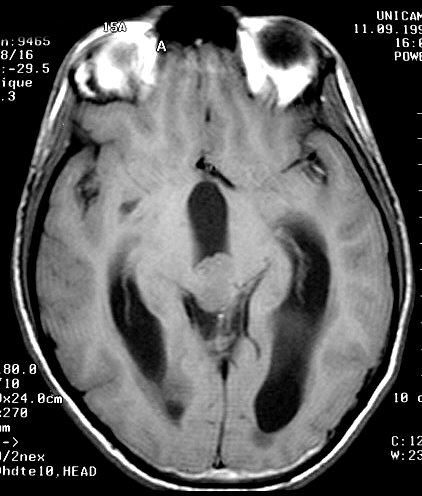
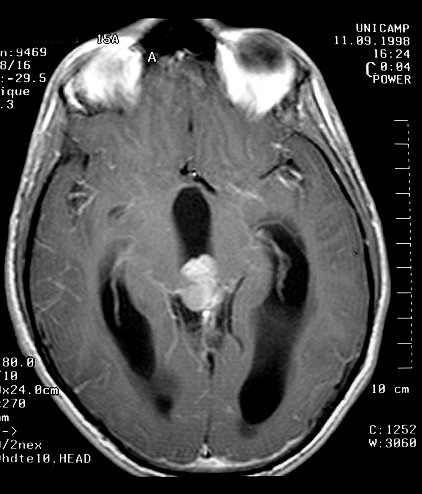
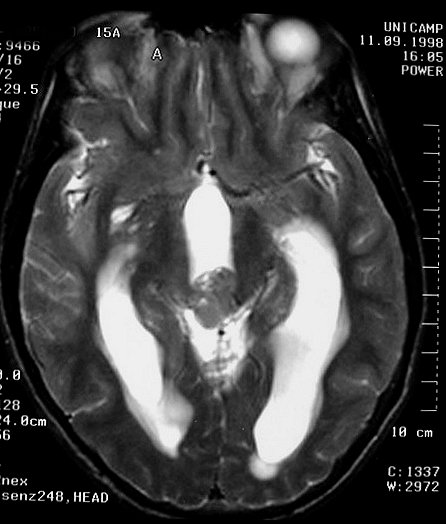
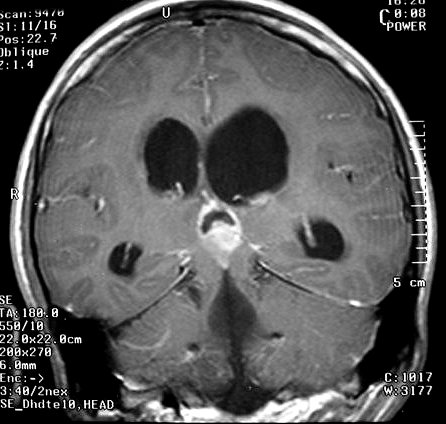
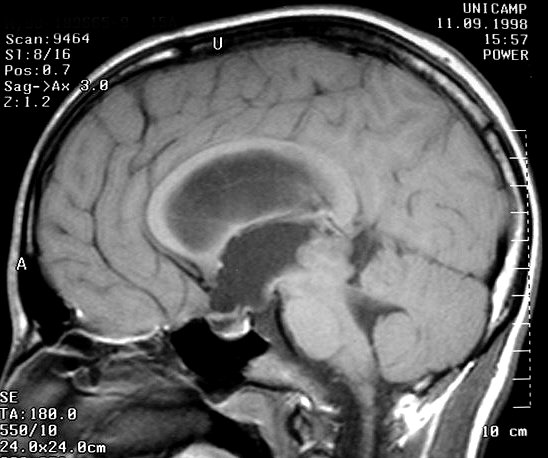
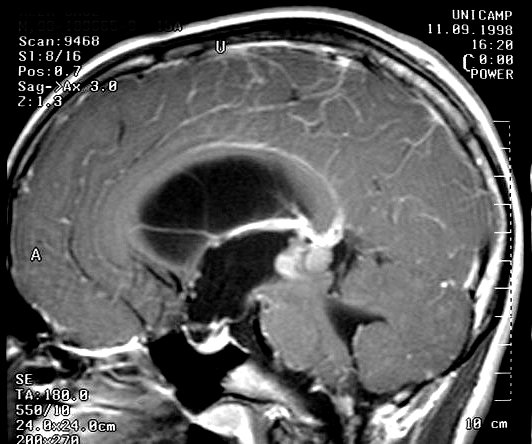
| Pineoblastoma. M. 15 a. A neoplasia é pequena, mas sua localização estratégica acima da lâmina quadrigêmea leva à compressão precoce do aqueduto cerebral e hidrocefalia. A boa delimitação sugere um tumor de baixo grau, mas há comprovação histológica de pineoblastoma. | |||
| TC com contraste | RM axial, T1 sem contraste | T1 com contraste | T2 |
 |
 |
 |
 |
| Coronal, T1 com contraste | Sagital, T1 sem contraste | T1 com contraste |
 |
 |
 |
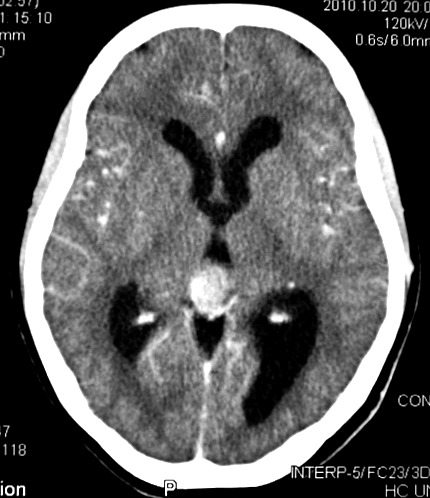
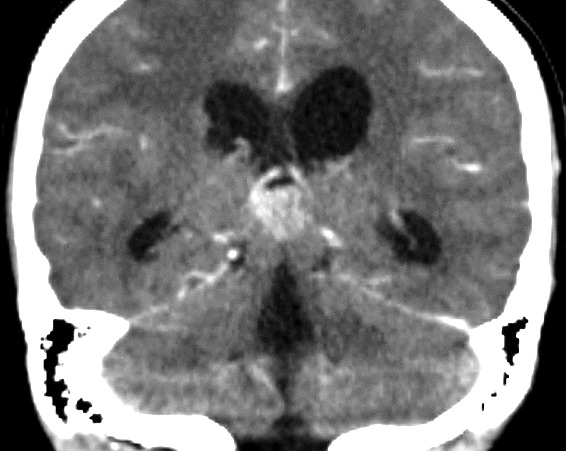
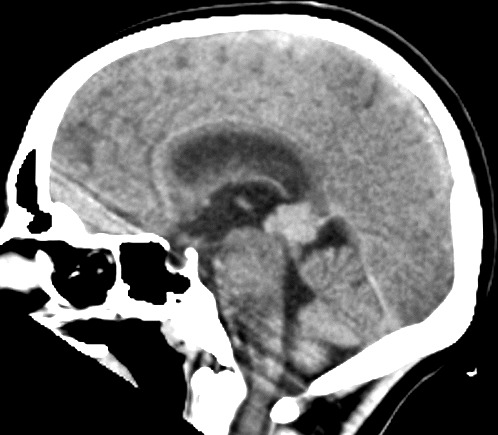
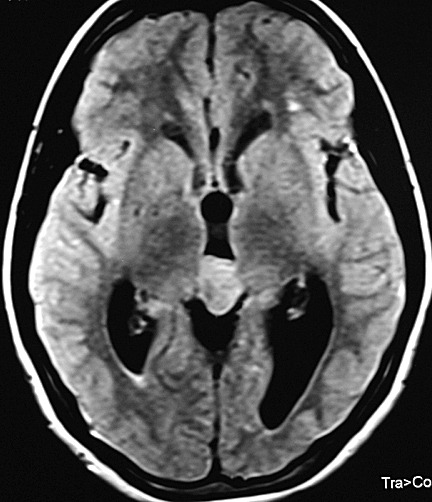
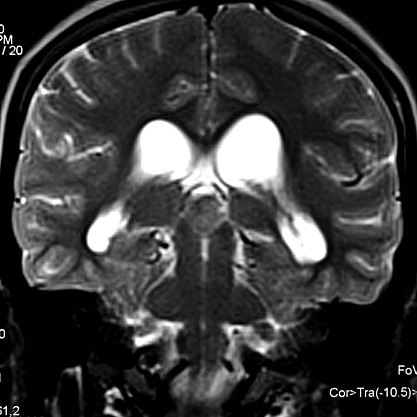
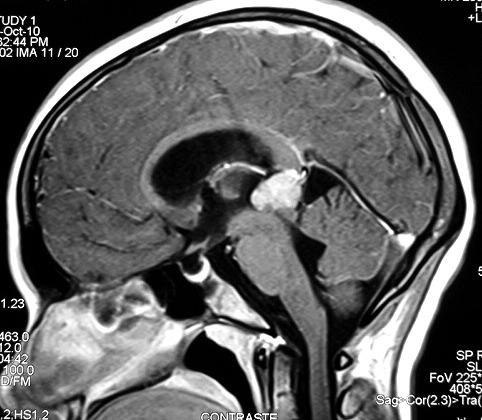
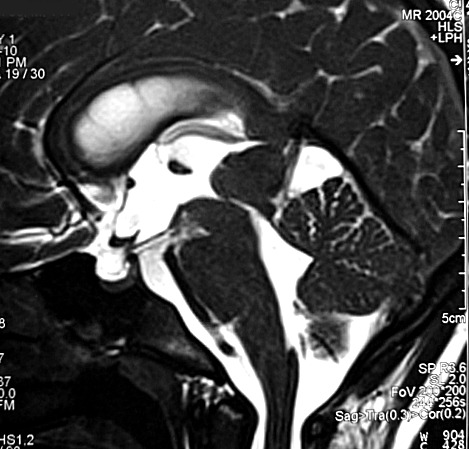
| Pineoblastoma, tomografia computadorizada. F. 14 a. As características de imagem são praticamente superponíveis às do caso acima. | |||
| Axial, com contraste | Coronal, com contraste | Sagital, sem contraste | Com contraste |
 |
 |
 |
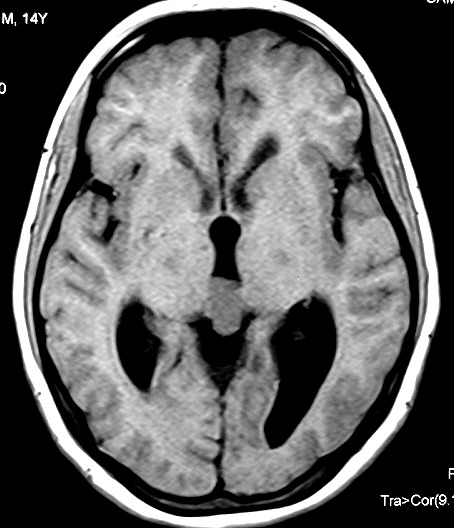
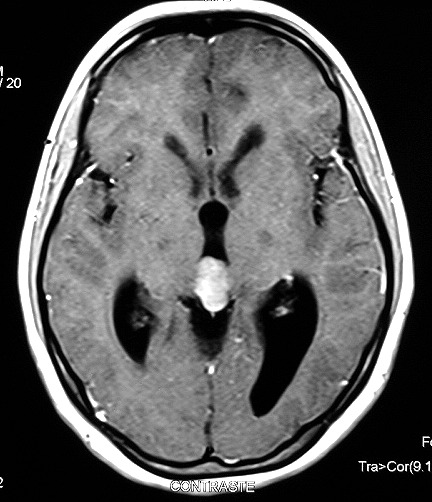
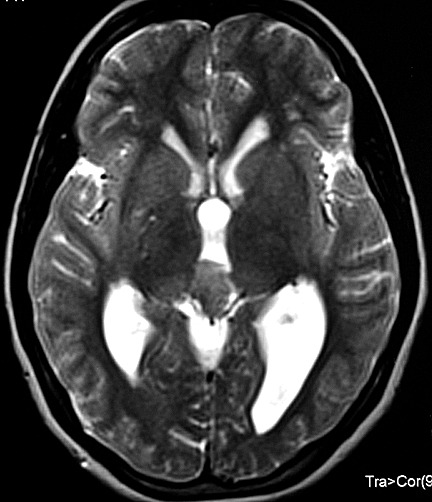
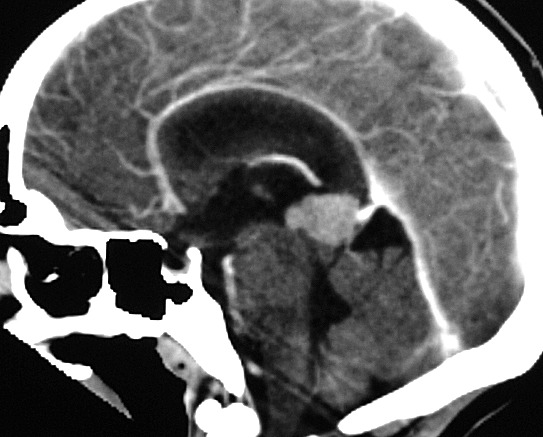 |
| RM axial, T1 sem contraste | T1 com contraste | T2 | FLAIR |
 |
 |
 |
 |
| Coronal, T2 | Sagital, T1 com contraste | T2 |
 |
 |
 |
| Para exame histológico e imunohistoquímico deste caso, clique. | ||
|
|
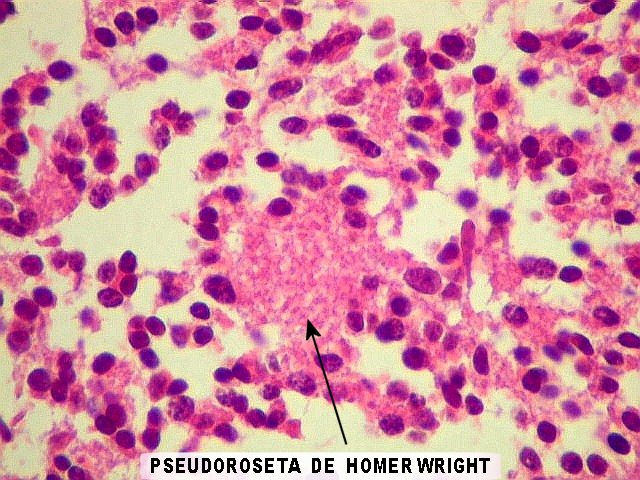
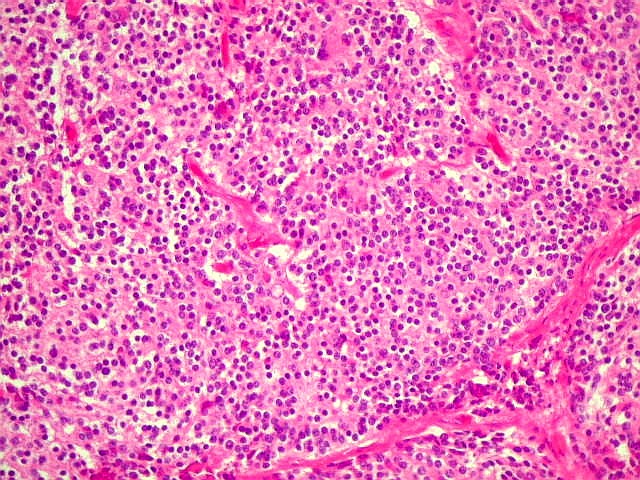
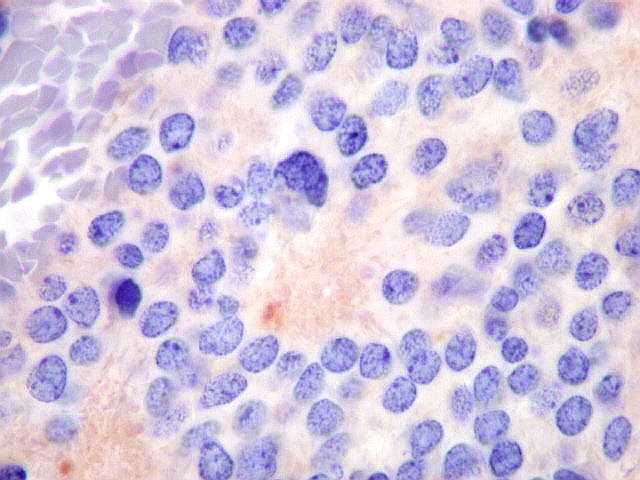
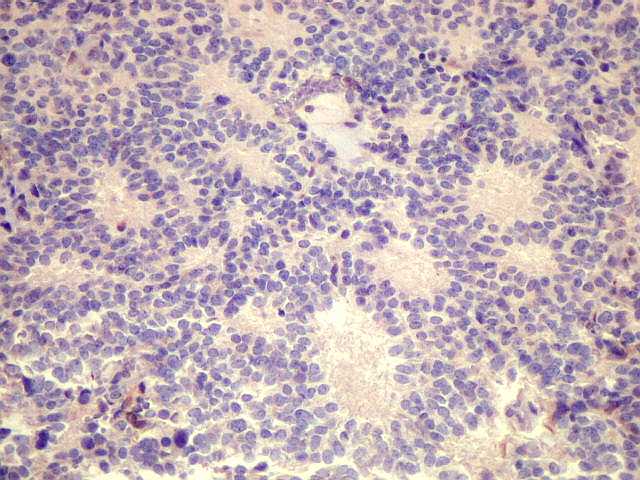
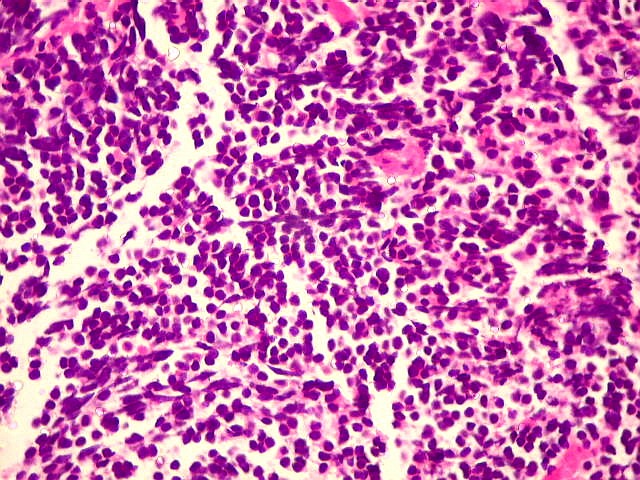
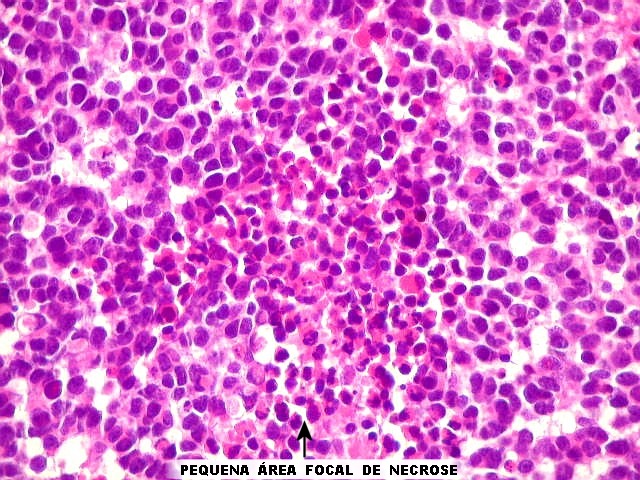
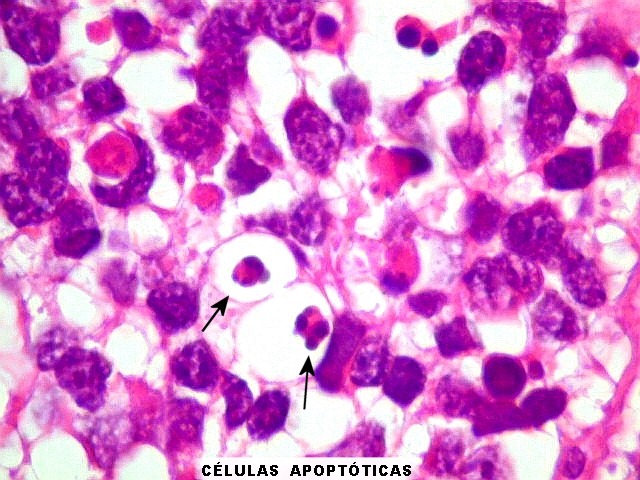
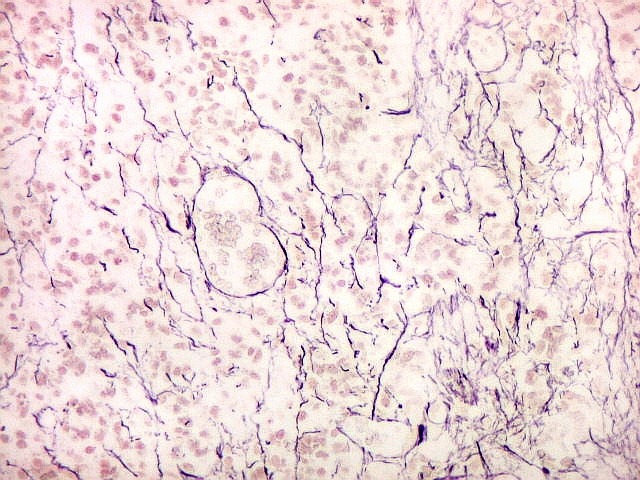
| Pineoblastoma - HE. Tumor de células primitivas, com as feições características de um PNET (tumor neuroectodérmico primitivo). As células têm núcleos hipercromáticos e compactos, relativamente regulares apesar do caráter maligno, e citoplasma muito escasso. O arranjo é sólido, não tendo sido observadas rosetas. Alguns septos conjuntivos, melhor evidenciados com tricrômico de Masson, abaixo, dividem o tumor em lóbulos. Há freqüentes células em apoptose, mas não se observam áreas extensas de necrose. | |
 |
 |
 |
 |
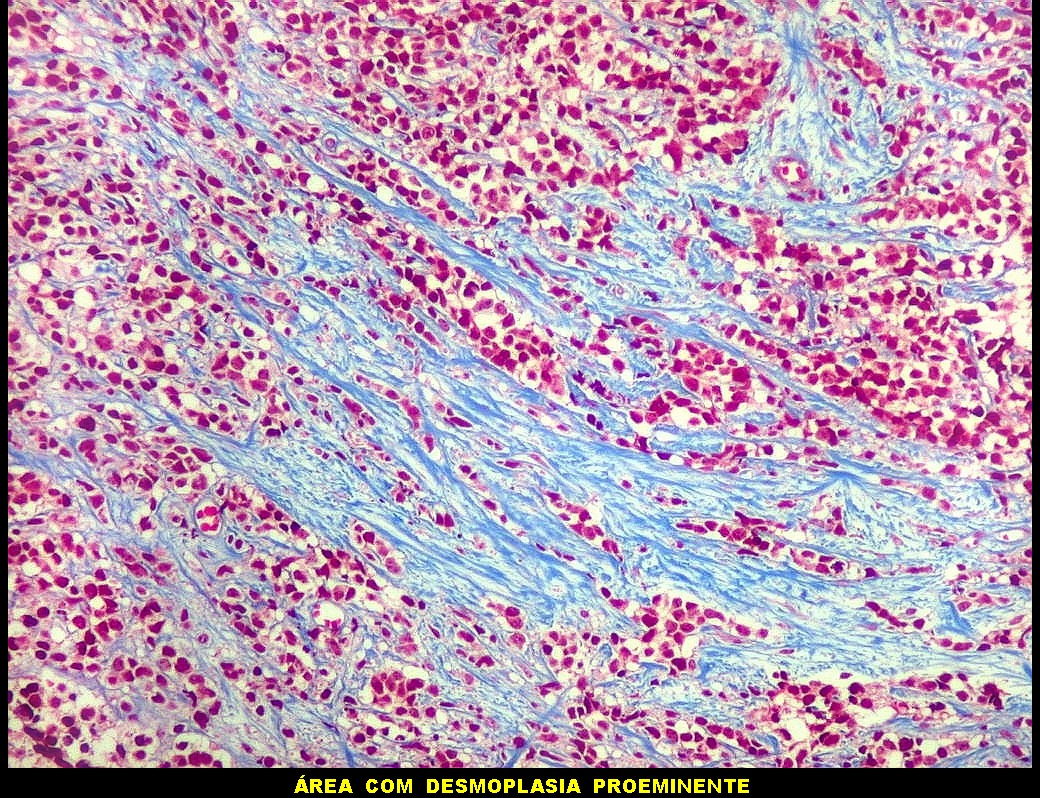
| Tricrômico de Masson. Fibras colágenas entre as células neoplásicas podem ser abundantes, dando caráter desmoplásico. | Reticulina. Fibras reticulínicas delicadas estão irregularmente distribuídas. Ilhotas livres de reticulina lembram a arquitetrua dos meduloblastomas desmoplásicos. |
 |
 |
|
|
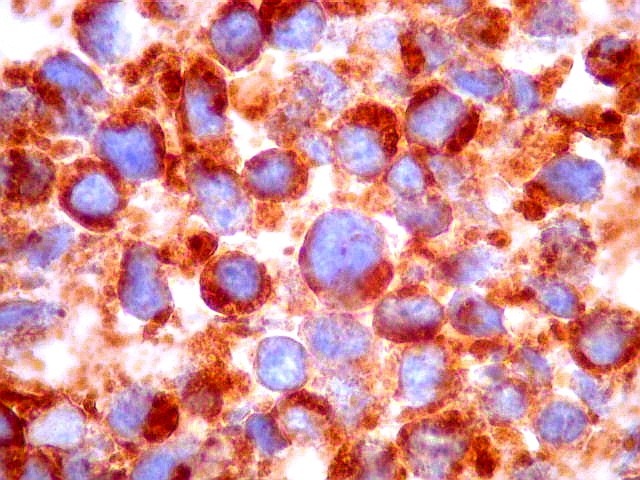
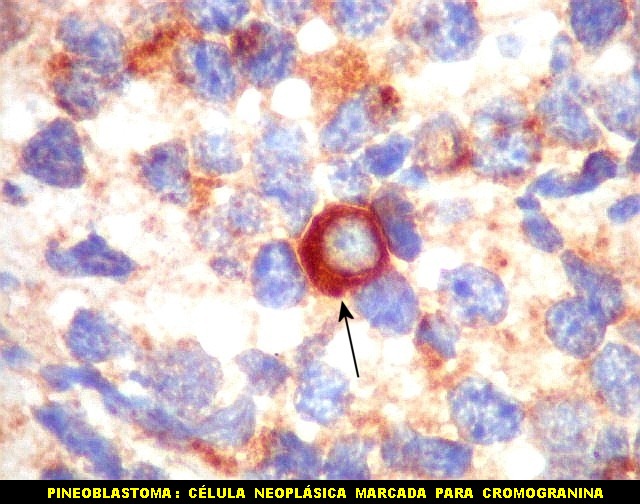
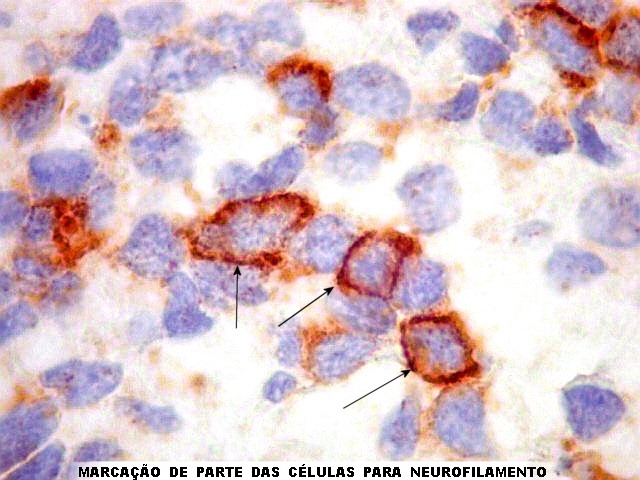
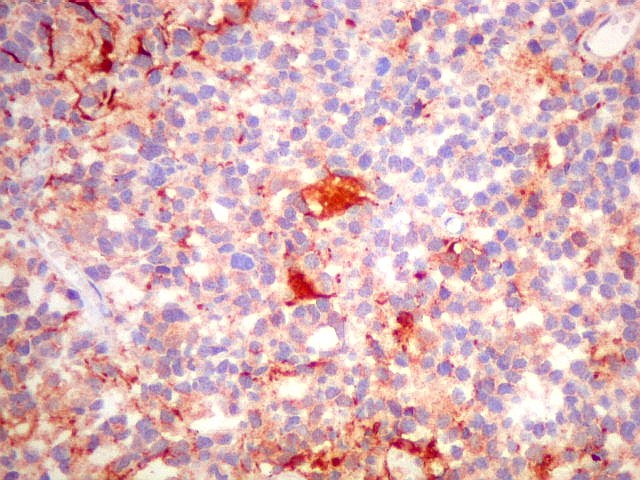
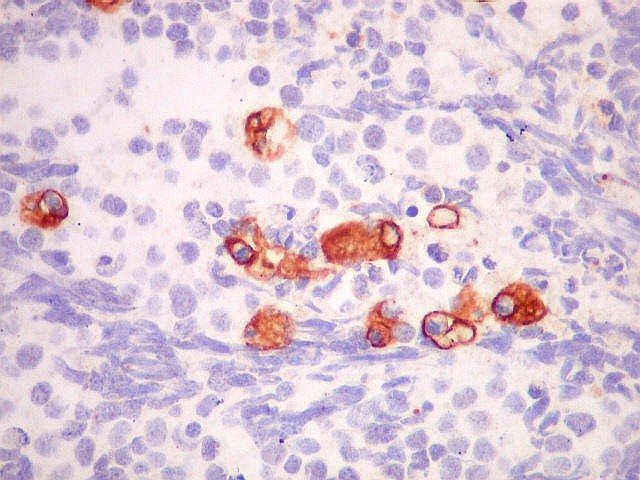
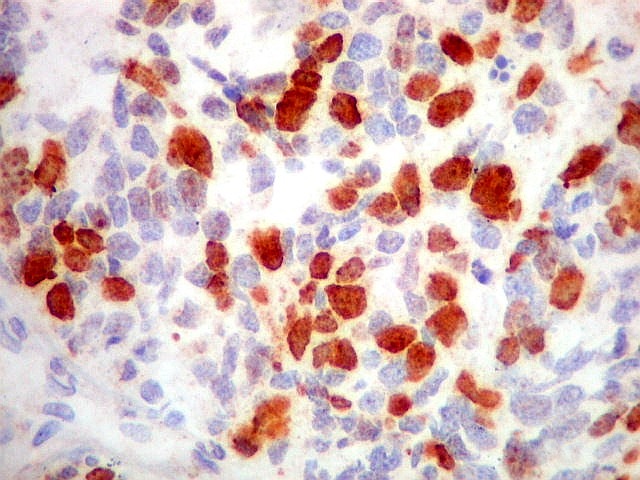
| Marcação citoplasmática para três antígenos neuronais: neurofilamento, sinaptofisina e cromogranina demonstra que as células neoplásicas, embora imaturas, recapitulam a natureza neuronal dos pineocitos, as células parenquimatosas da pineal. Há positividade em padrão membrana para CD56. Antígenos gliais são de modo geral negativos, exceto em raras células (possível diferenciação divergente). Vimentina é negativa nas células do tumor e positiva em outros tipos de células, como macrófagos, fibroblastos e vasos. Ki-67 é alto, indicando intensa atividade proliferativa. p53 foi positivo só em raros núcleos. | ||
| SNF | Cromogranina | NF |
 |
 |
 |
| CD56 | GFAP | S-100 |
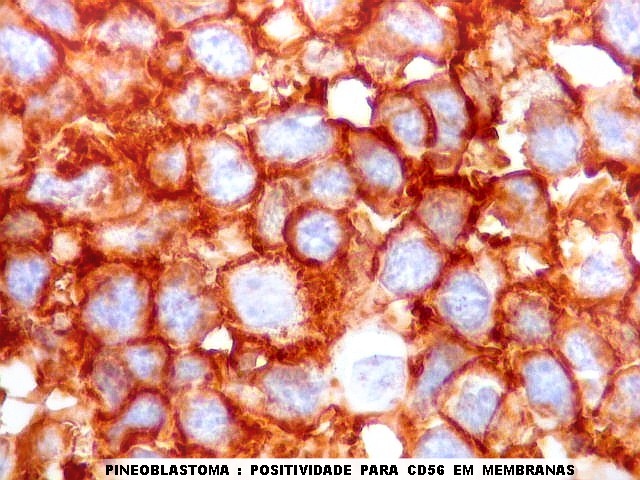 |
 |
 |
| VIM | Ki-67 | p53 |
 |
 |
 |
| Tumores germinativos_: | Neuroimagem | Neuropatologia | Tumores do parênquima pineal : | Neuroimagem | Neuropatologia |
| Banco de imagens : | Pineal normal,
HE e imunohistoquímica |
Tumores da pineal | Textos ilustrados linkados: | Tumores germinativos do SNC | Tumores do parênquima pineal |
|
|
| Neuropatologia
- Graduação |
Neuropatologia -
Estudos de casos |
Neuroimagem
- Graduação |
Neuroimagem -
Estudos de Casos |
Roteiro
de aulas |
Textos
de apoio |
Correlação
Neuropatologia - Neuroimagem |
| Índice alfabético - Neuro | Adições recentes | Banco de imagens - Neuro | Textos ilustrados | Neuromuscular | Patologia - outros aparelhos | Pages in English |
|
|
|
|