|
|
|
|
|
|
|
|
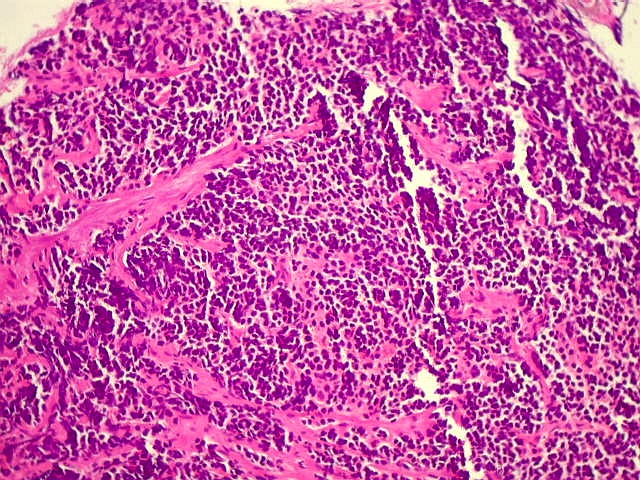
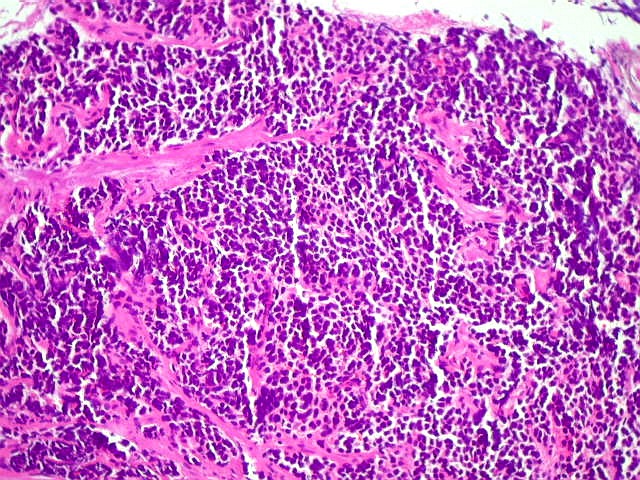
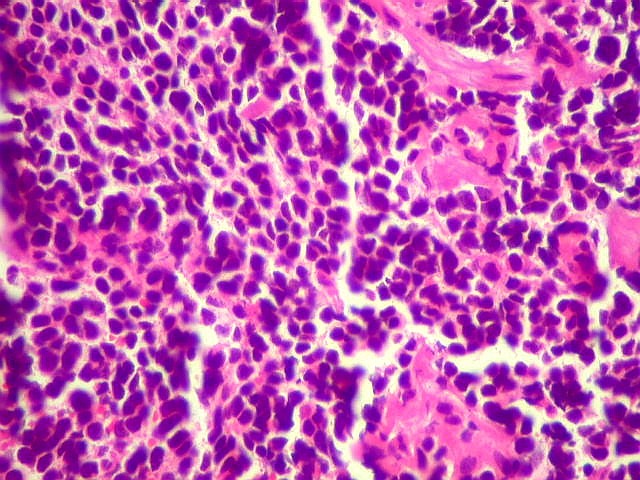
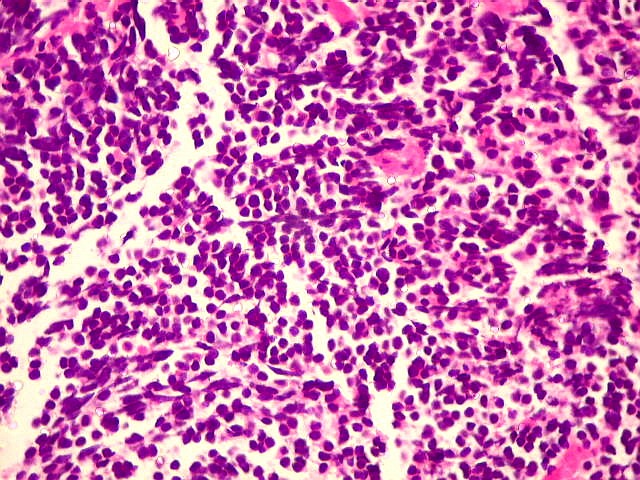
| Pineoblastoma.
Espécime
proveniente da região pineal de paciente feminina, 20 anos. Para
acompanhamento da lesão em exames de imagem entre 1996 e 2003, clique.
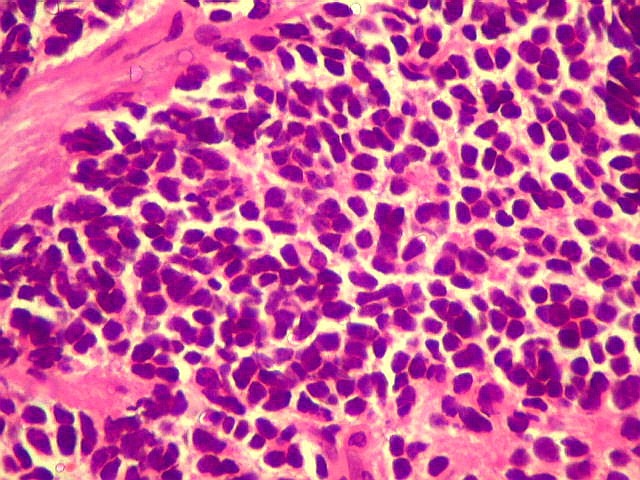
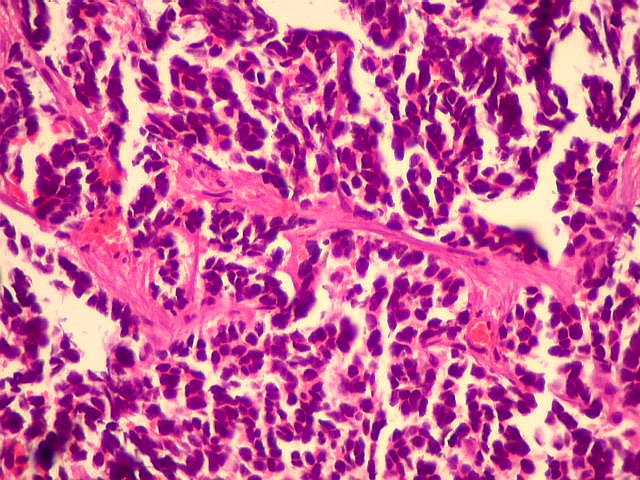
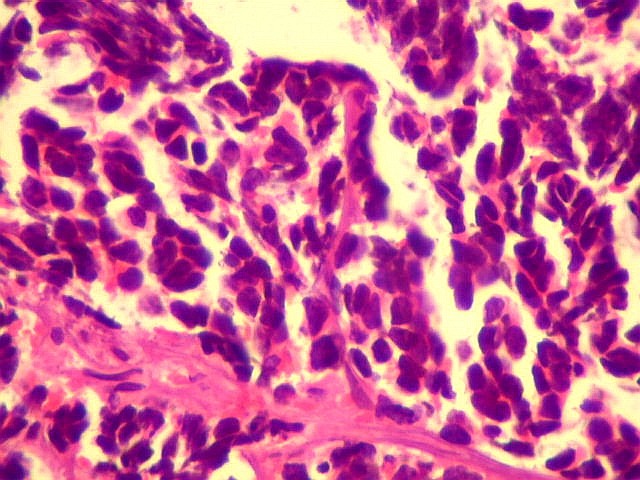
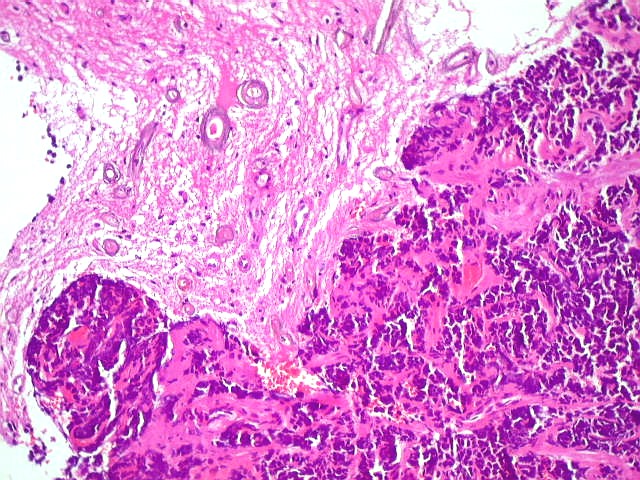
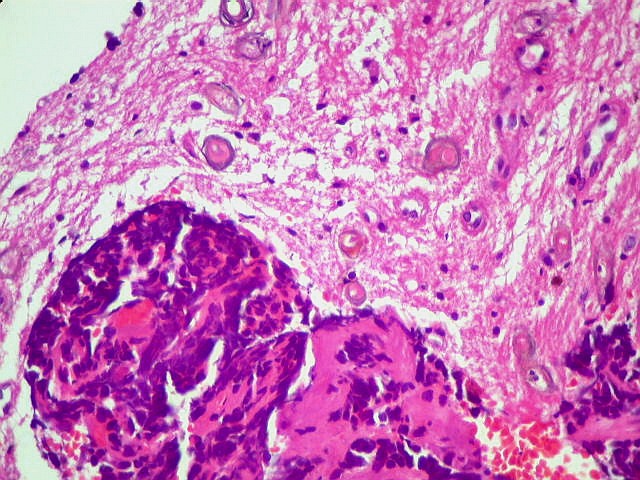
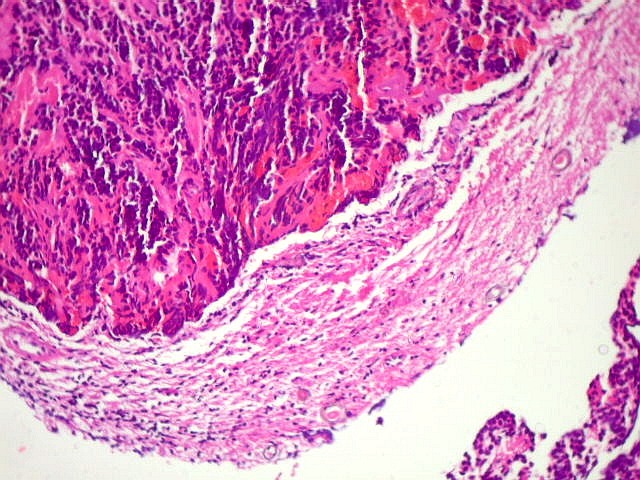
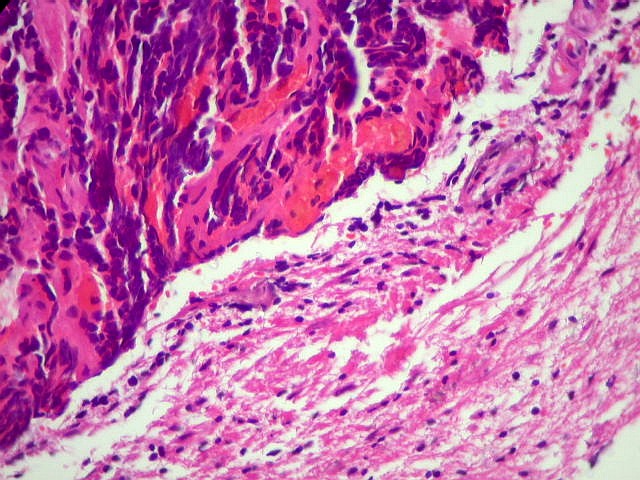
A lâmina corada por HE mostra tumor indiferenciado, constituído por pequenas células regulares, com núcleo hipercromático e citoplasma escasso, dispostas em agrupamentos sólidos entre traves conjuntivo-vasculares. O aspecto geral é o de um tumor neuroectodérmico primitivo ou PNET. As células não formam rosetas do tipo Flexner-Wintersteiner (com centro vazio) nem pseudorosetas do tipo Homer Wright (com centro preenchido por prolongamentos celulares), nem rosetas do tipo pineocitomatoso. Nesta amostra não foram vistas mitoses ou necrose. O tumor é bem delimitado do tecido nervoso na pequena área onde esta avaliação foi possível. |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
| PINEOBLASTOMAS
e outros PNETs supratentoriais.
Definição. Os pineoblastomas são tumores malignos originados no parênquima da glândula pineal e são considerados um tumor neuroectodérmico primitivo ou PNET. O termo PNET pode ser usado como sinônimo de tumor embrionário (de pequenas células) do SNC sem sinais de diferenciação ou apresentando diferenciação divergente, ao longo de linhagens neuronal, astrocitária, ependimária, muscular ou melânica. Os PNETs mais freqüentes são os meduloblastomas, exclusivos do cerebelo. Localização. PNETs são raros no compartimento supratentorial, onde incluem, além dos pineoblastomas, os neuroblastomas centrais, os ependimoblastomas e os retinoblastomas. Faltam estatísticas precisas sobre a incidência. Se considerados todos os tumores pediátricos com características histológicas de PNET, até 15% têm localização supratentorial (incluídos os hemisférios cerebrais, região supraselar e região pineal). Se os exemplos na região pineal (pineoblastomas) são excluídos, este número cai para 5.5%. Incidência. A faixa etária para pineoblastomas é de 6 meses a 16 anos (média 8.6 anos). Para os PNETs cerebrais e supraselares é 4 semanas a 10 anos (média 5.5 anos). A razão entre sexo masculino e feminino para os pineoblastomas é de 1:1.5 (para PNETs cerebrais e supraselares é 2:1). Clínica. PNETs cerebrais apresentam-se com hipertensão intracraniana, convulsões, distúrbios da consciência ou déficits localizados. Os supraselares produzem distúrbios visuais ou endócrinos. Os da pineal causam obstrução das vias liquóricas e desordens do movimento ocular. O pineoblastoma às vezes associa-se ao retinoblastoma bilateral, chamando-se o complexo de síndrome do retinoblastoma trilateral. O componente intracraniano desta tríade geralmente origina-se na pineal, mas pode ocorrer também na região selar. Imagem e macroscopia. Os tumores variam de tamanho, sendo os supraselares e pineais menores que os dos hemisférios cerebrais. Estes podem ser grandes, com ou sem cistos ou hemorragias. Demarcação entre o tumor e o cérebro pode ser indistinta ou nítida. A cor é rósea-avermelhada. São moles ou, se contêm componente desmoplásico, podem ser firmes. 50 a 70% têm calcificações (inclusive nos pineais). Edema em volta dos tumores geralmente não é intenso. Nos métodos de imagem, os PNETs em geral, inclusive os pineoblastomas, captam contraste. Microscopia. As feições são as mesmas do meduloblastoma, com células pouco diferenciadas ou indiferenciadas em arranjo sólido. Pode haver rosetas de Homer Wright, canais ependimários e rosetas de Flexner-Wintersteiner (de centro vazio), estas particularmente em tumores da pineal. As rosetas de Flexner são mais próprias dos retinoblastomas. Seu encontro em pineoblastomas não deve causar surpresa, em vista da função fotoreceptora da pineal em animais inferiores. PNETs podem ter estroma fibroso. O índice mitótico é geralmente alto. Todos PNETs são classificados histologicamente como grau IV de malignidade segundo a OMS. Imunohistoquímica. Pode haver diferenciação divergente para neurônios, astrócitos e epêndima. Os anticorpos mais úteis são GFAP (diferenciação astrocitária), e NSE, NF e SNF (diferenciação neuronal). Prognóstico. Pineoblastomas tendem a recidivar logo e disseminar-se pelo espaço subaracnóideo. O prognóstico é pior abaixo dos 2 anos, seja para PNETs pineais ou outros supratentoriais. Crianças com PNETs supratentoriais não-pineais têm sobrevida de 5 anos de 34%. Para meduloblastomas, este índice hoje é de 85%. Fontes: Burger PC, Scheithauer BW. Tumors of the Central Nervous System. AFIP Atlas of Tumor Pathology. Armed Forces Institute of Pathology, Washington, DC, 1994. Lantos PL, Vandenberg SR, Kleihues P. Tumours of the Nervous System. Chapter 9 in Greenfield's Neuropathology, 6th Ed, Graham DI, Lantos PL (eds). Arnold, London, 1997. Mena H et al. Pineoblastoma. in Kleihues P, Cavenee WK (eds) Tumours of the Nervous System. Pathology and Genetics. World Health Organization (WHO) and International Agency for Research on Cancer (IARC). IARCPress, Lyon, 2000. |
| Caso contribuído pelo Dr. Marcelo Alvarenga, Campinas, SP. |
| Para TC, RM desta paciente, clique » | 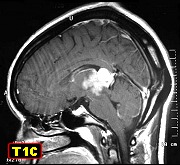 |
| Tumores germinativos_: | Neuroimagem | Neuropatologia | Tumores do parênquima pineal : | Neuroimagem | Neuropatologia |
| Banco de imagens : | Pineal normal,
HE e imunohistoquímica |
Tumores da pineal | Textos ilustrados linkados: | Tumores germinativos do SNC | Tumores do parênquima pineal |
|
|
| Neuropatologia
- Graduação |
Neuropatologia -
Estudos de casos |
Neuroimagem
- Graduação |
Neuroimagem -
Estudos de Casos |
Roteiro
de aulas |
Textos
de apoio |
Correlação
Neuropatologia - Neuroimagem |
| Índice alfabético - Neuro | Adições recentes | Banco de imagens - Neuro | Textos ilustrados | Neuromuscular | Patologia - outros aparelhos | Pages in English |
|
|