|
|
|
|
|
|
|
|
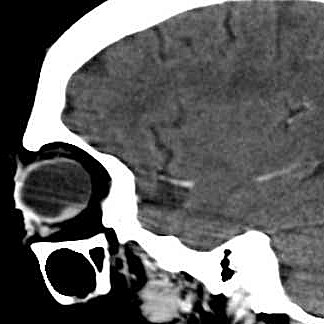
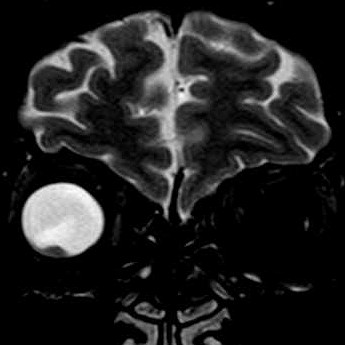
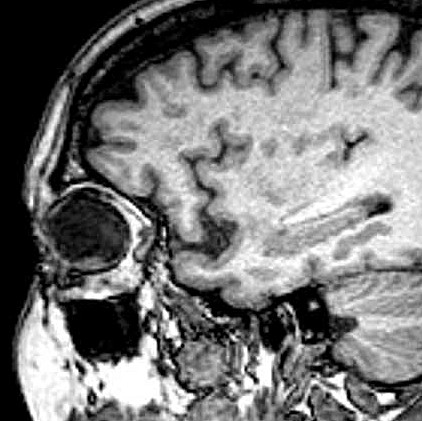
| A paciente, com 67 anos na época desta cirurgia, já havia sido enucleada do olho esquerdo aproximadamente com a idade de 25 anos. A pobreza de informações no prontuário não permite avançar sobre antecedentes que tenham motivado as alterações oculares. Não há, nos dados clínicos ou nos exames de imagem, evidência de síndromes genéticas associadas a neoplasias de vias ópticas, como a esclerose tuberosa e a neurofibromatose do tipo 1. Por isso, o tumor do olho direito foi considerado provavelmente esporádico. A enucleação foi motivada pela suspeita de lesão melânica ou metastática, sendo a paciente já totalmente amaurótica. Para TC, RM, clique. Texto sobre astrocitomas intraoculares. |
| Destaques da microscopia. | ||
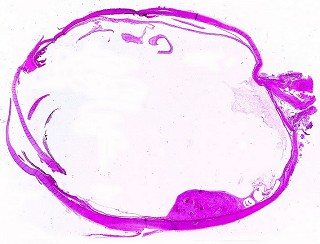
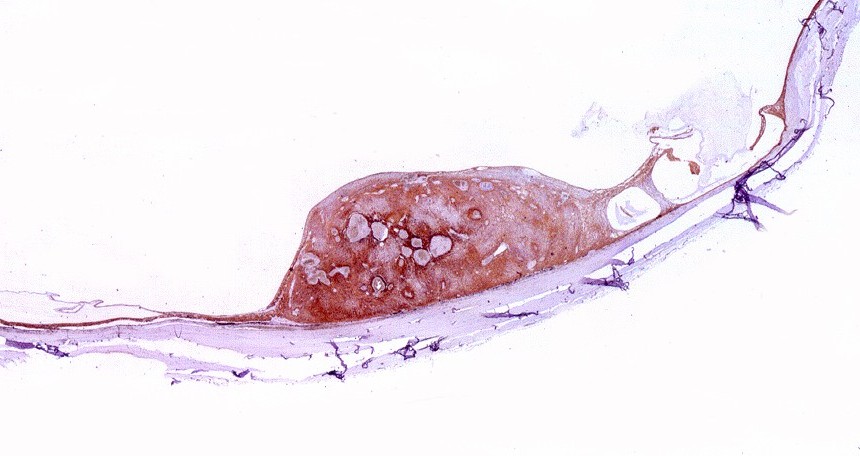
| HE. Lâmina escaneada mostra pequeno tumor retiniano não invasivo. | Astrocitoma pilocítico - células alongadas com prolongamentos paralelos | Corpos hialinos granulosos |
 |
 |
 |
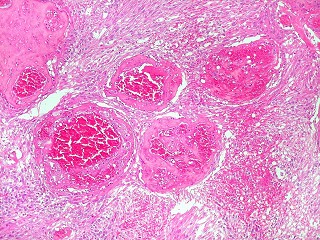
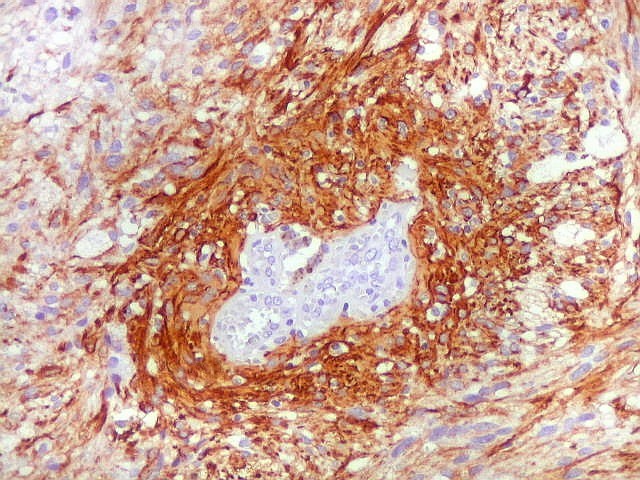
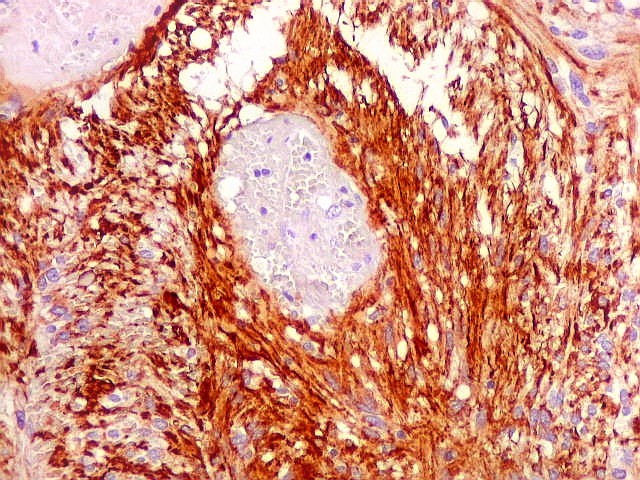
| Vasos espessados, dilatados, hialinizados | GFAP. Lâmina escaneada, tumor GFAP positivo. | GFAP, aspecto geral em aumento fraco |
 |
 |
 |
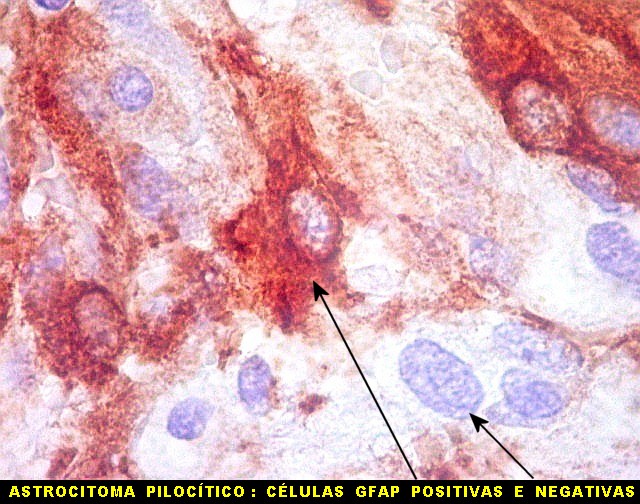
| Caráter pilocítico das células neoplásicas, positivas para GFAP. | Positividade variável das células neoplásicas, algumas totalmente negativas | Vasos negativos, circundados por coroa de células positivas |
 |
 |
 |
| VIM. Positividade difusa no tumor e no endotélio vascular | S-100. Positividade nuclear e citoplasmática em parte das células neoplásicas | Ki-67. Positividade em cerca de 1-3 % dos núcleos das células neoplásicas. |
 |
 |
 |
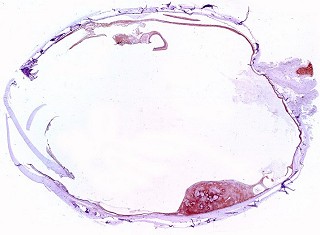
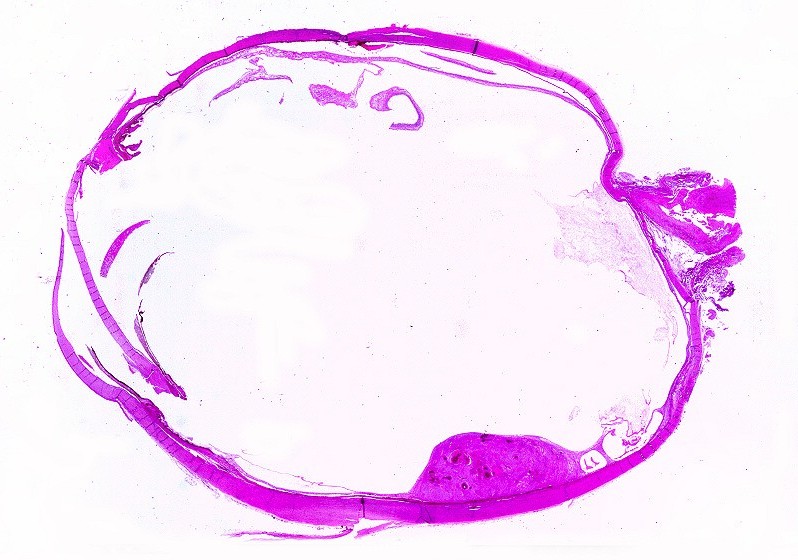
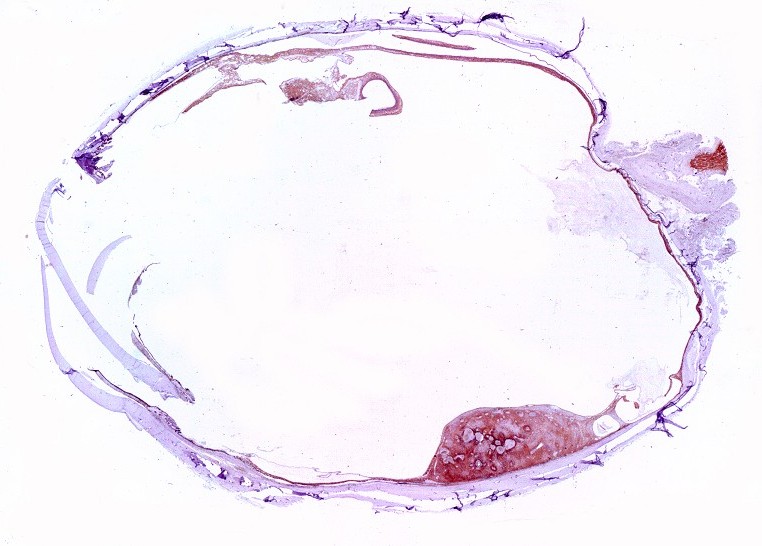
| Lâmina
escaneada.
Corte em plano sagital pelo globo ocular, incluindo o pequeno tumor na parte inferior da retina documentado em TC e RM. Lesão não invade a esclera. Resolução 600 dpi (dots per inch). Abaixo, 1200 dpi. |
 |
 |
 |
 |
 |
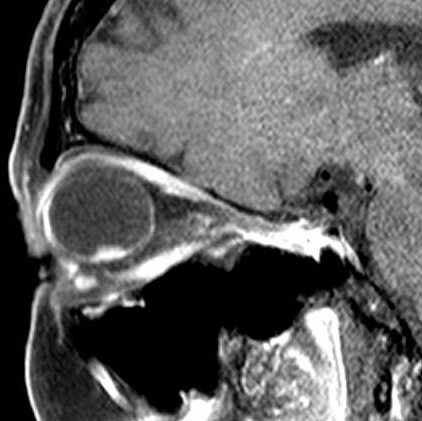 |
| TC sagital com contraste | RM coronal T2 | RM sagital sem contraste | Com contraste |
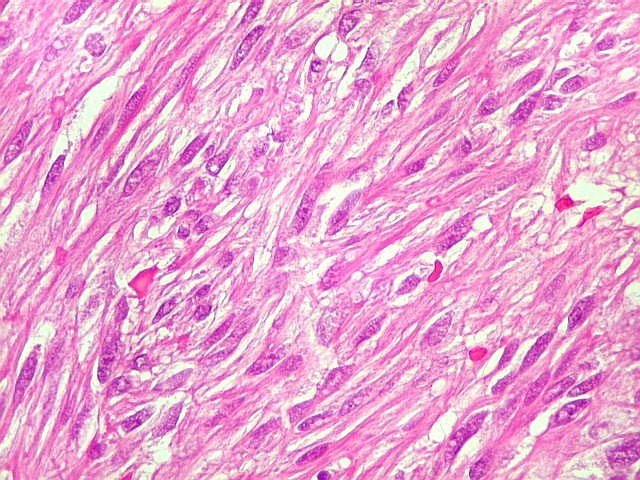
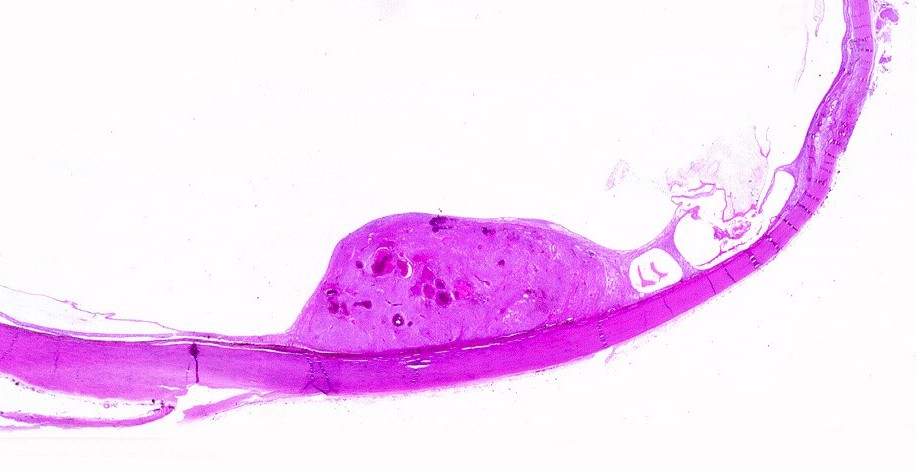
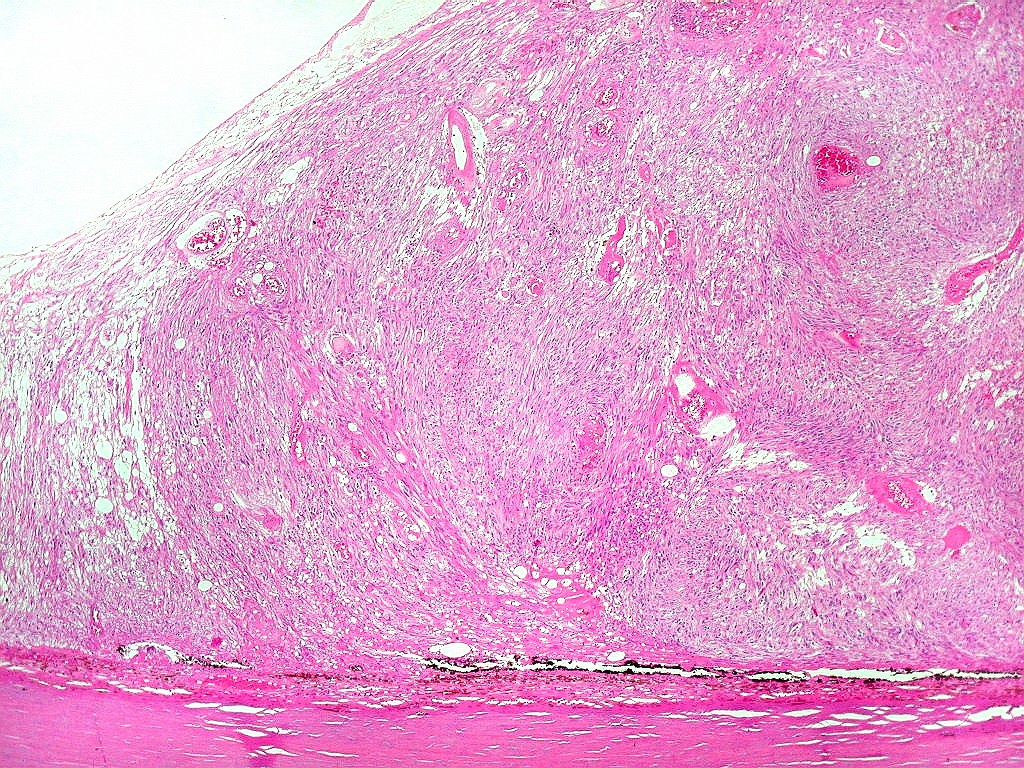
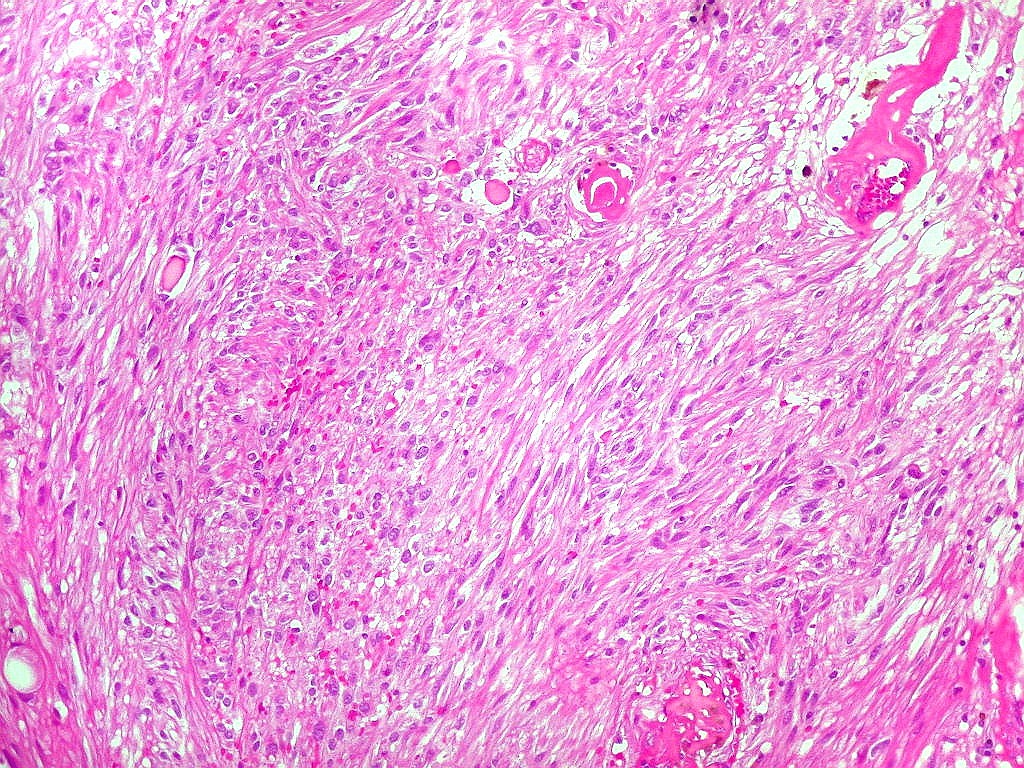
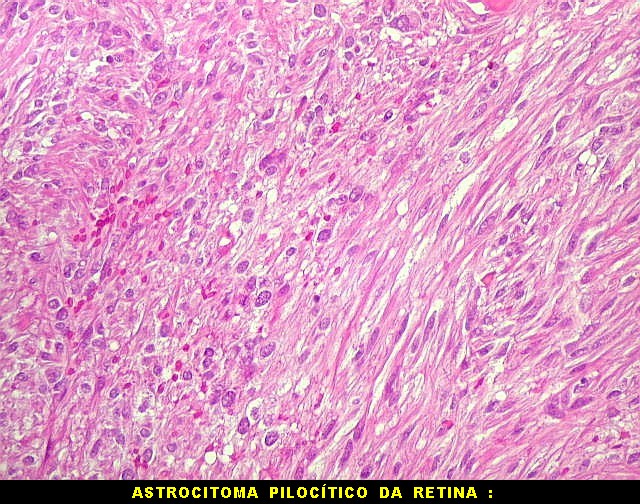
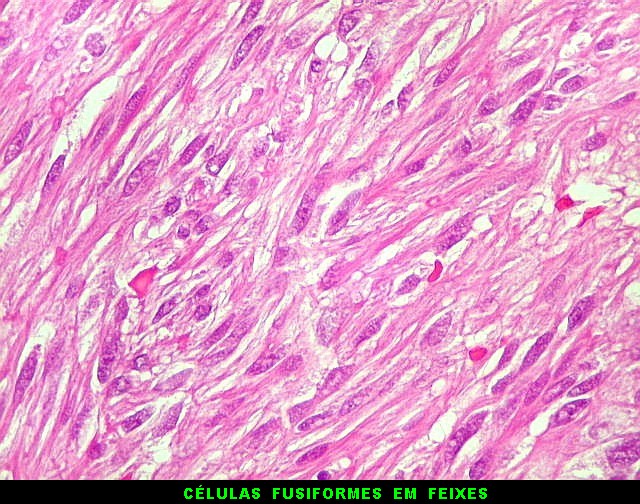
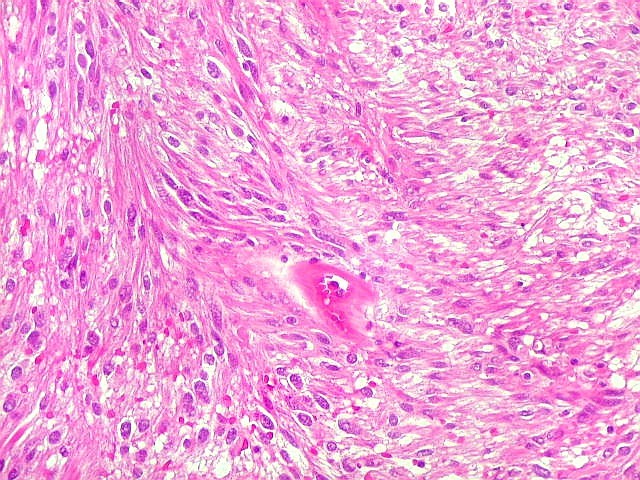
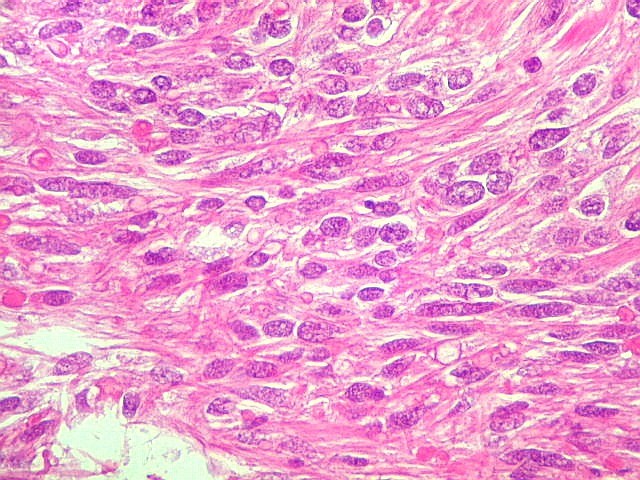
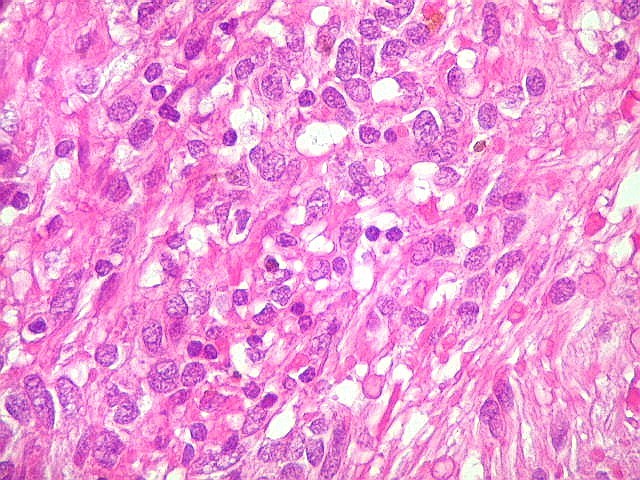
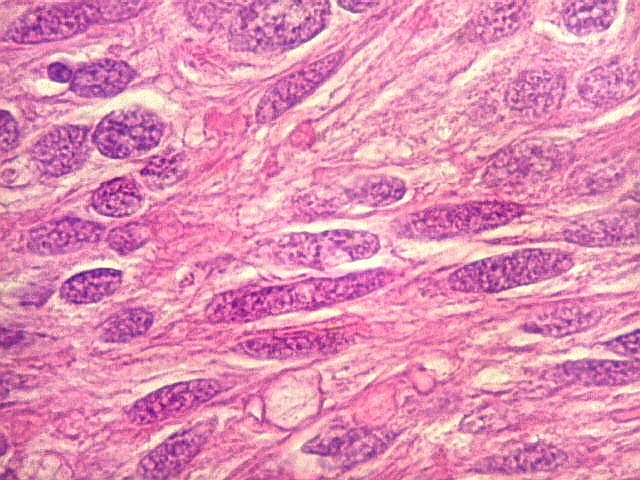
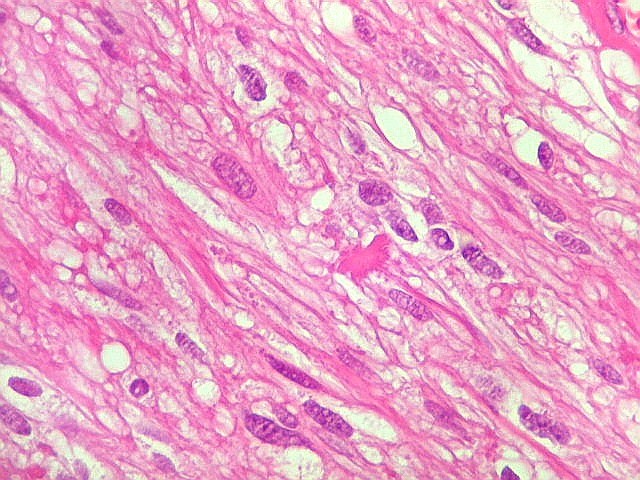
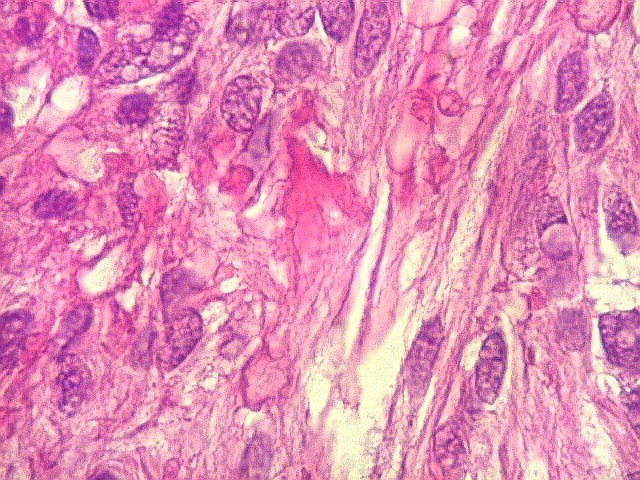
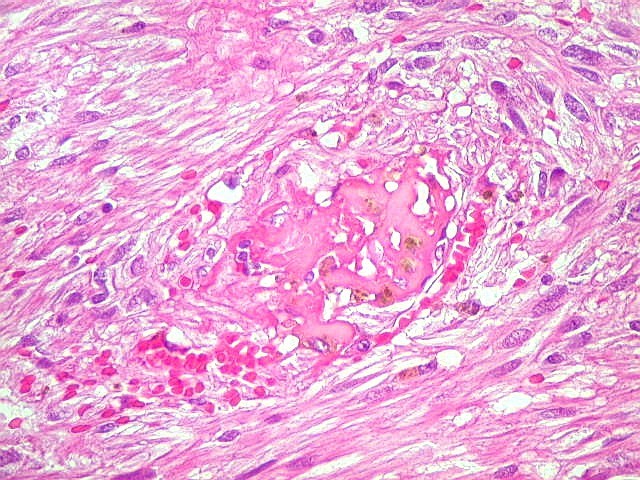
| HE, aspecto geral. O tumor é constituído por células alongadas ou fusiformes, arranjadas em feixes multidirecionados. A natureza astrocitária das mesmas é documentada pela positividade para GFAP. O caráter fusocelular é próprio dos astrocitomas pilocíticos, assim como corpos hialinos granulosos. Vasos anômalos, dilatados e com trombose também são observados em astrocitomas pilocíticos. A neoplasia é bem delimitada, restrita à retina, a qual está totalmente atrófica. Não há infiltração da coróide, esclera ou componente extraocular. Abaixo, detalhes das extremidades do tumor. |
 |
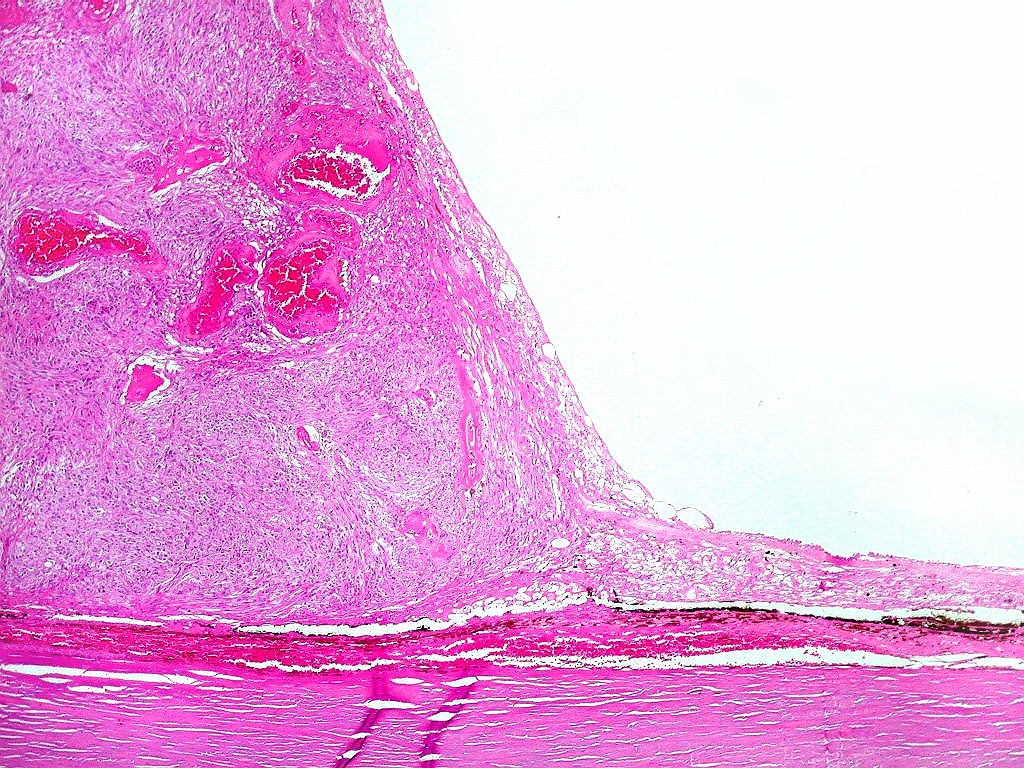 |
| Astrocitoma pilocítico. Textura fasciculada, células arranjadas em feixes que se entrelaçam em várias direções. Os núcleos têm cromatina fina e bem distribuída, o citoplasma é róseo e de limites imprecisos. Não há atipias nucleares significativas. Mitoses ou necrose não foram observadas. |
 |
 |
 |
 |
 |
 |
 |
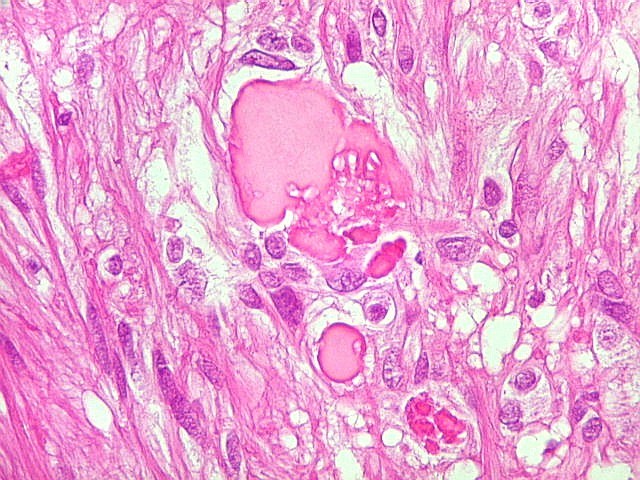
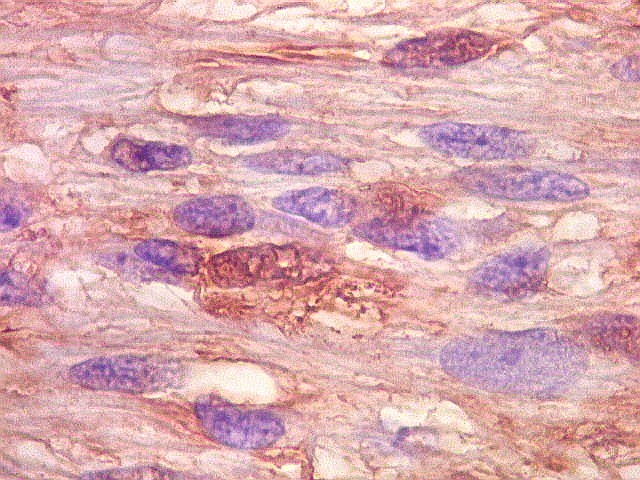
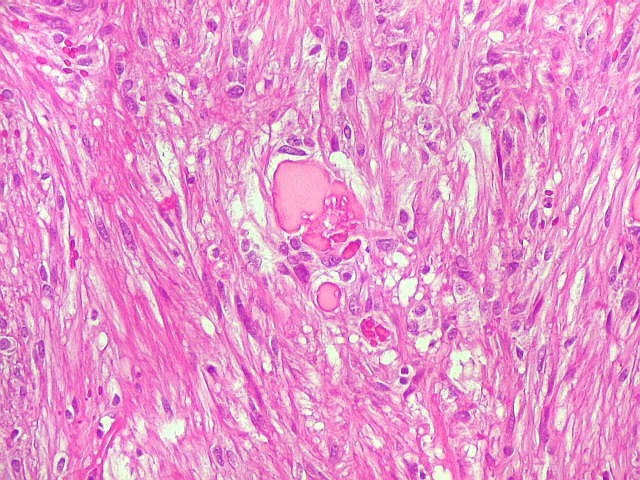
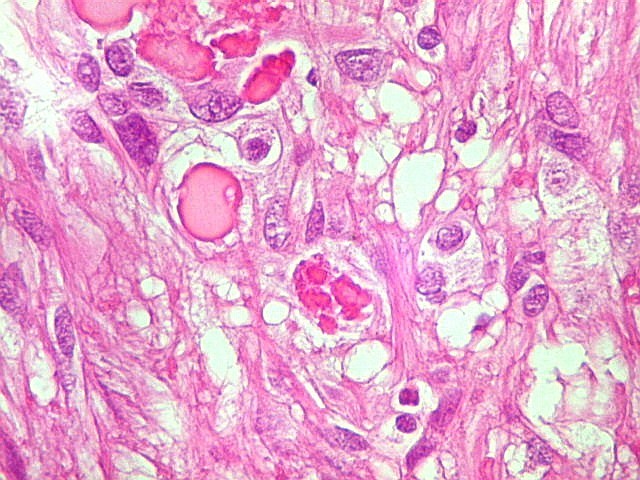
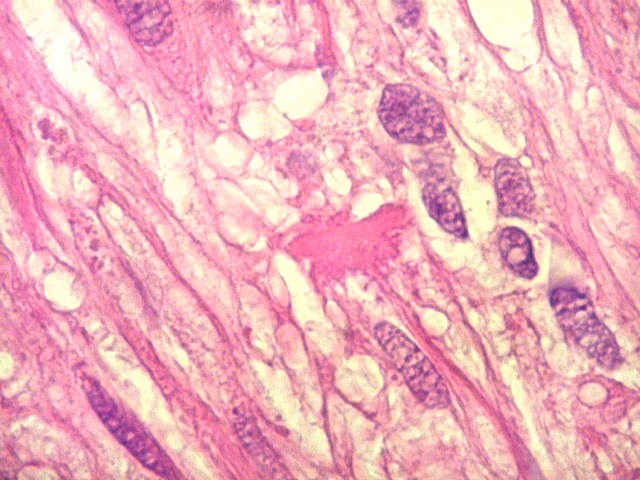
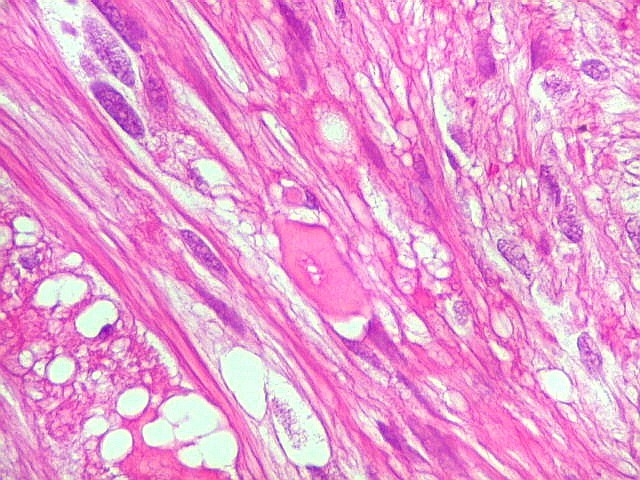
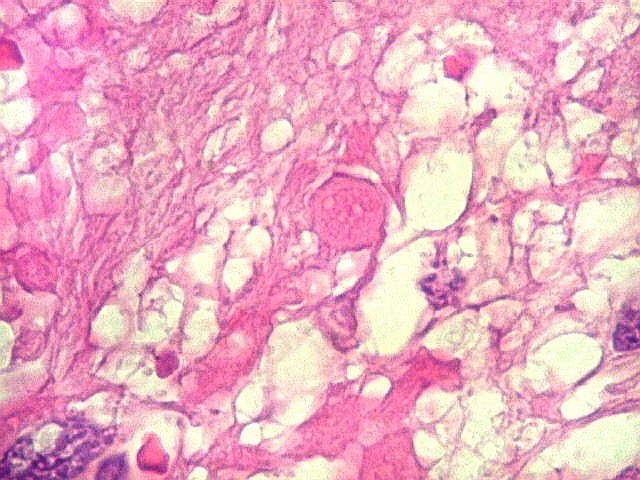
| Corpos hialinos eosinófilos. Numerosos corpos hialinos, homogêneos e eosinófilos, foram encontrados. Alguns tinham textura granulosa. Fibras de Rosenthal clássicas, porém, características dos astrocitomas pilocíticos, não foram observadas. Corpos hialinos indicam neoplasia glial de baixo grau histológico e são encontrados, além dos astrocitomas pilocíticos (1)(2), em gangliogliomas (1)(2)(3)(4), xantoastrocitomas (1)(2), e em gliose do tipo pilocítico nas proximidades de tumores de lento crescimento, como craniofaringiomas. | |
 |
|
 |
 |
 |
 |
 |
 |
 |
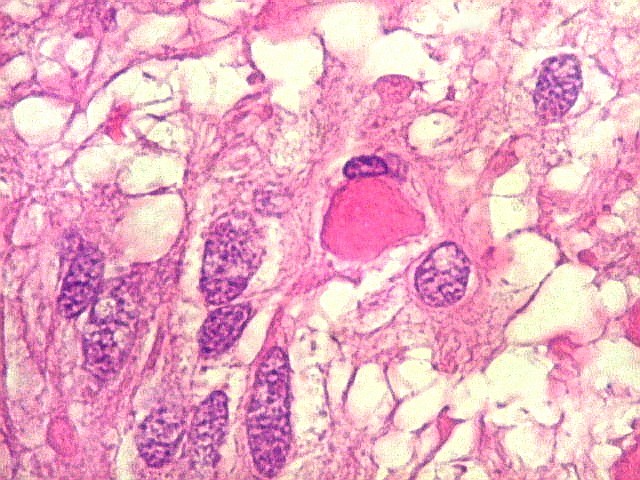 |
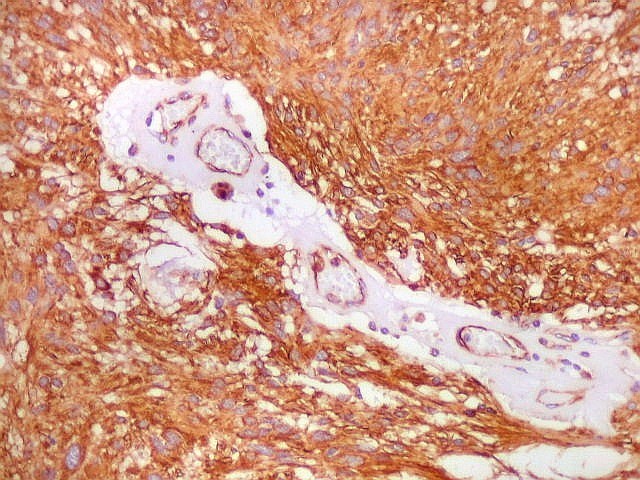
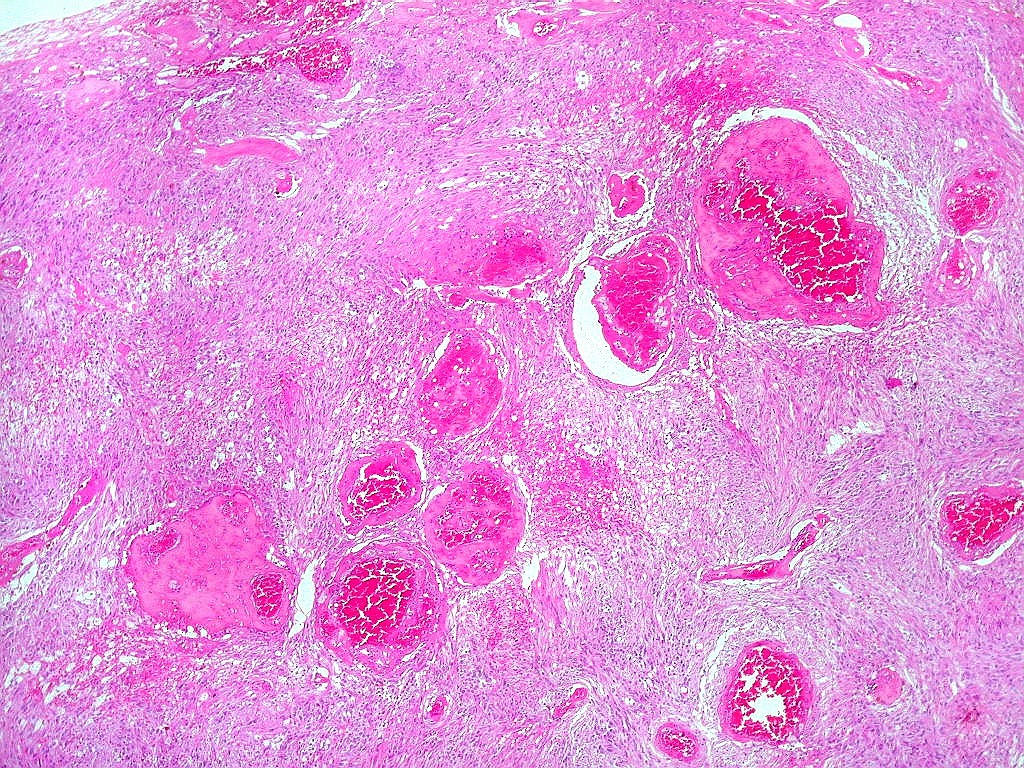
| Vasos. O tumor mostrava vasos anômalos, tortuosos, dilatados, com trombose. Não foi observada proliferação capilar (1)(2)(3) em guirlandas ou pseudoglomérulos, como não é raro nos astrocitomas pilocíticos. As lesões vasculares não pareciam acompanhar-se de repercussão no tecido tumoral, como necrose ou hemorragia. |
 |
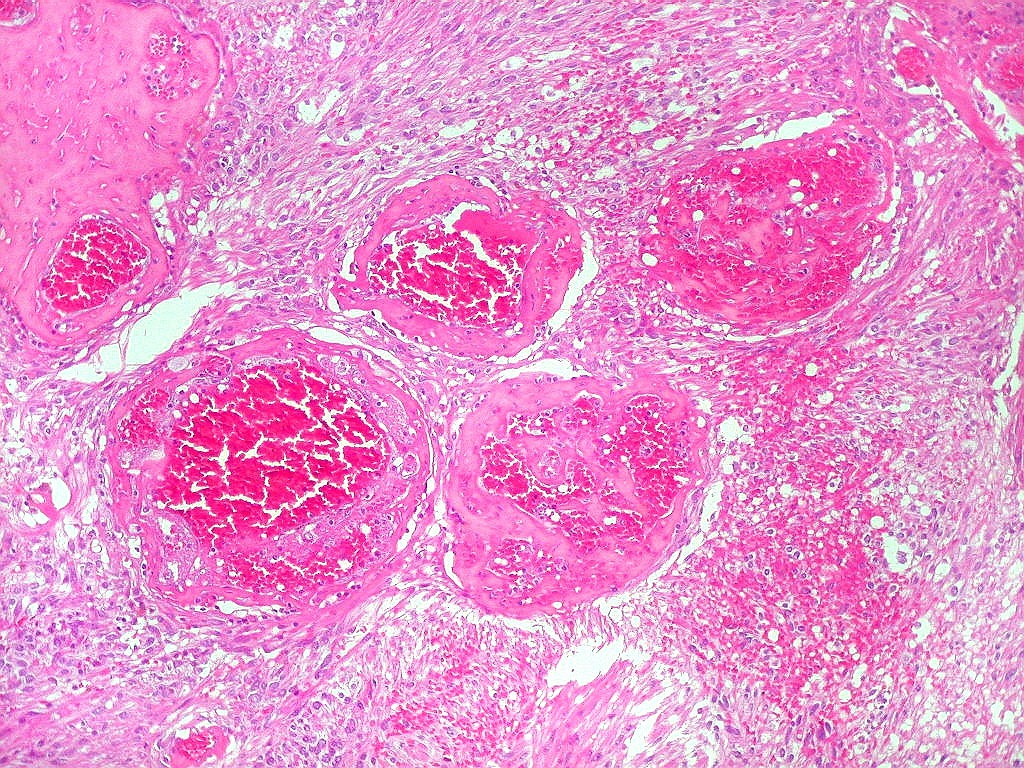 |
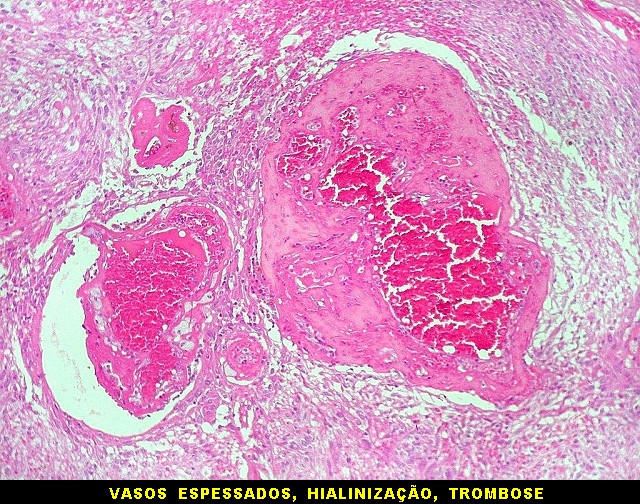 |
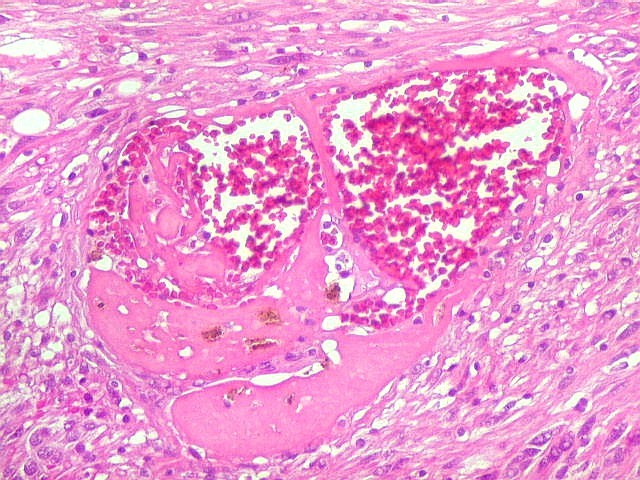 |
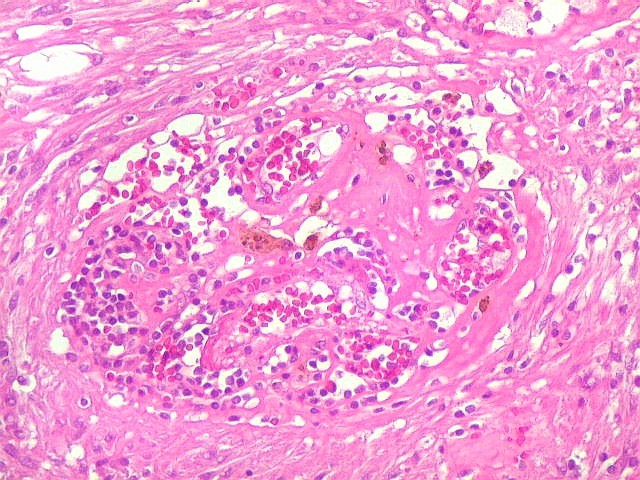 |
 |
|
|
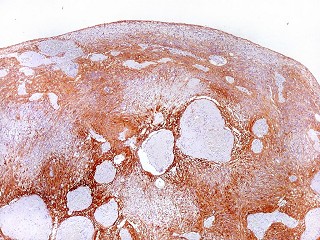
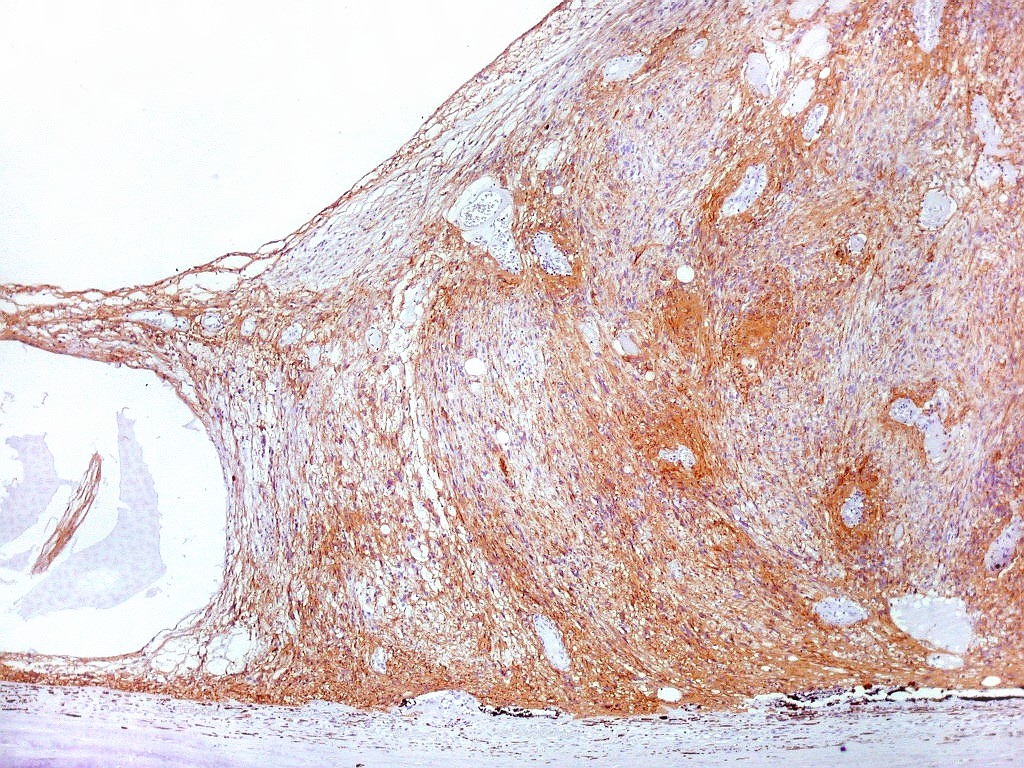
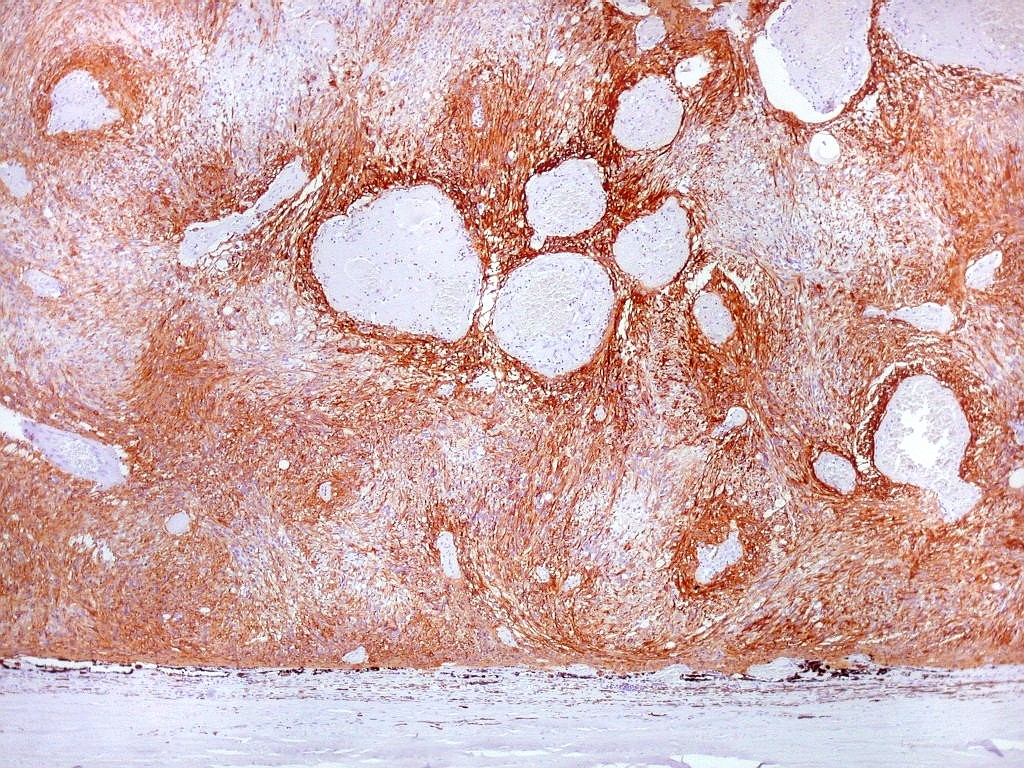
| GFAP.
Lâmina escaneada.
O pequeno tumor é rico em proteína glial ácida fibrilar, testemunhando sua natureza astrocitária. Esta imagem escaneada com 600 dpi (dots per inch). Abaixo, com 1200 dpi. |
 |
 |
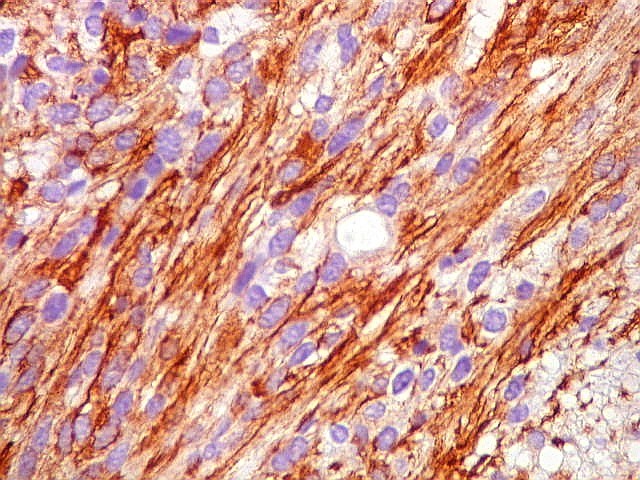
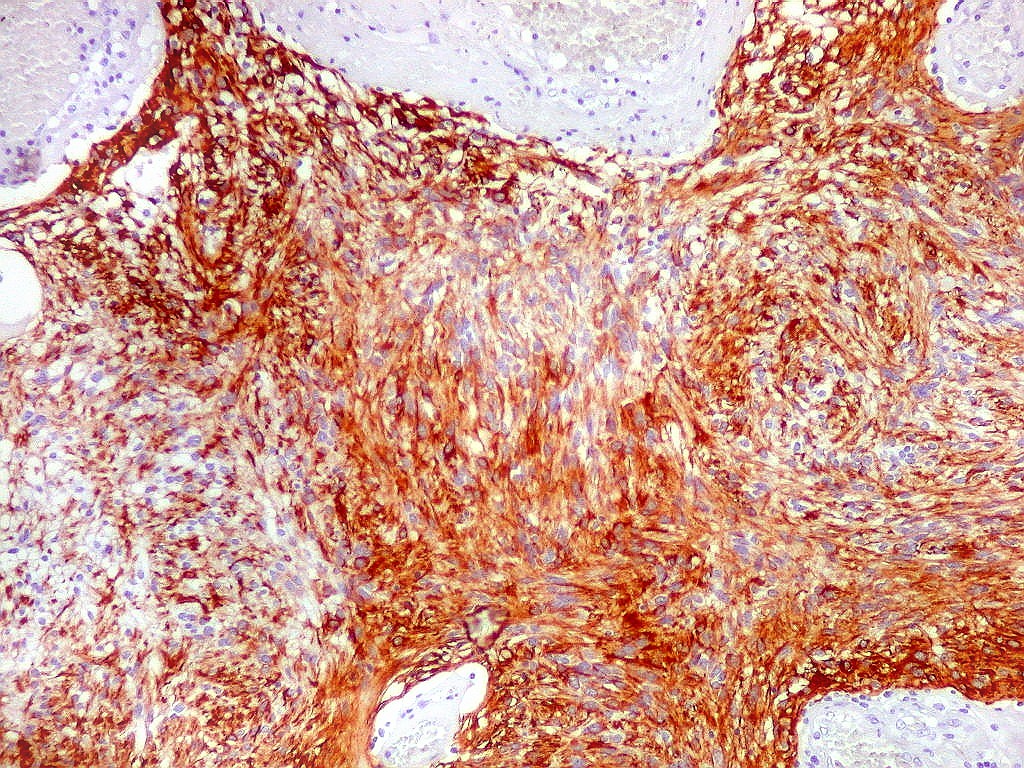
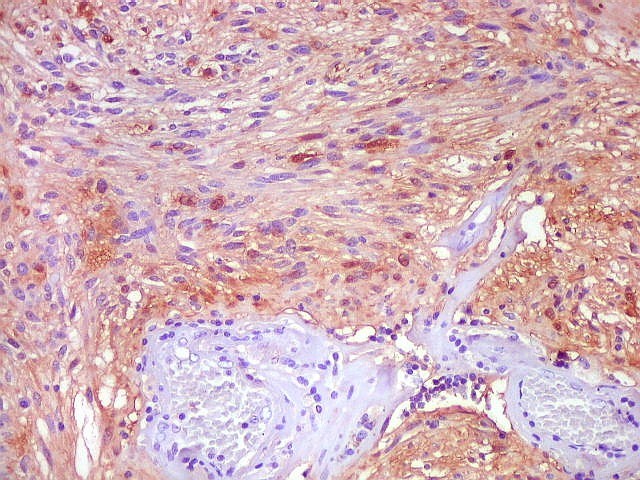
| GFAP. Aumento fraco. As fotos abaixo ilustram a positividade variável do tumor para GFAP (proteína glial ácida fibrilar ou glial fibrillary acidic protein), um filamento intermediário próprio de astrócitos. Há áreas e células fortemente positivas, outras praticamente negativas, demonstrando a variabilidade de expressão gênica, mesmo em células próximas da mesma neoplasia. Os vasos destacam-se pela negatividade. |
 |
 |
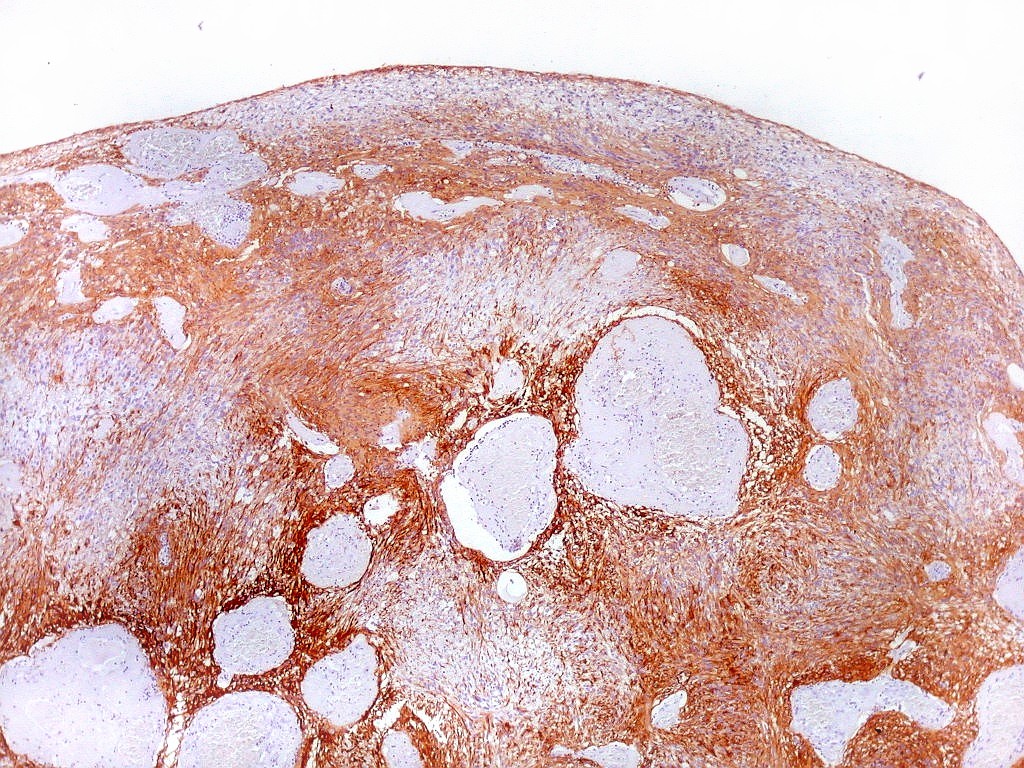 |
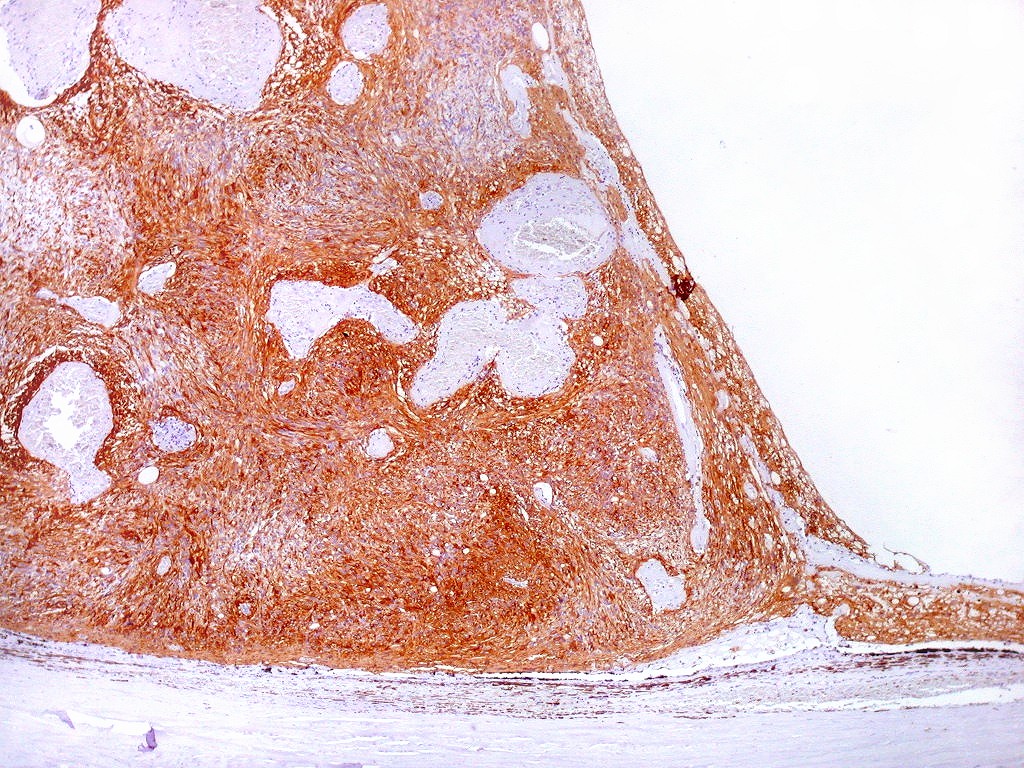 |
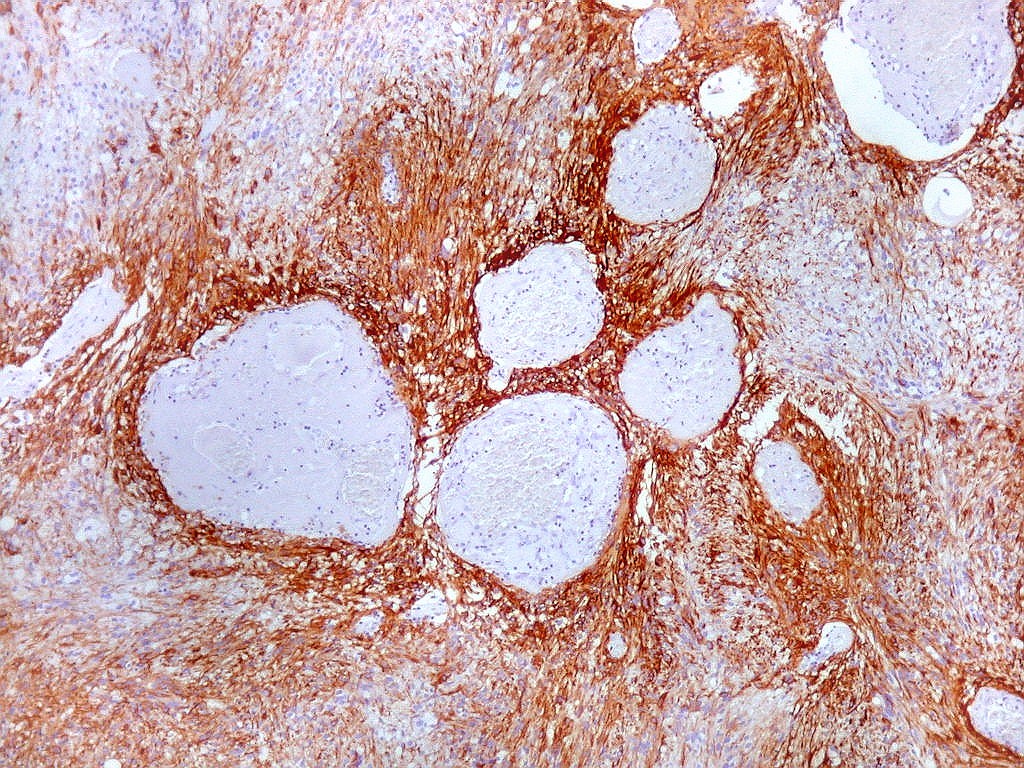 |
 |
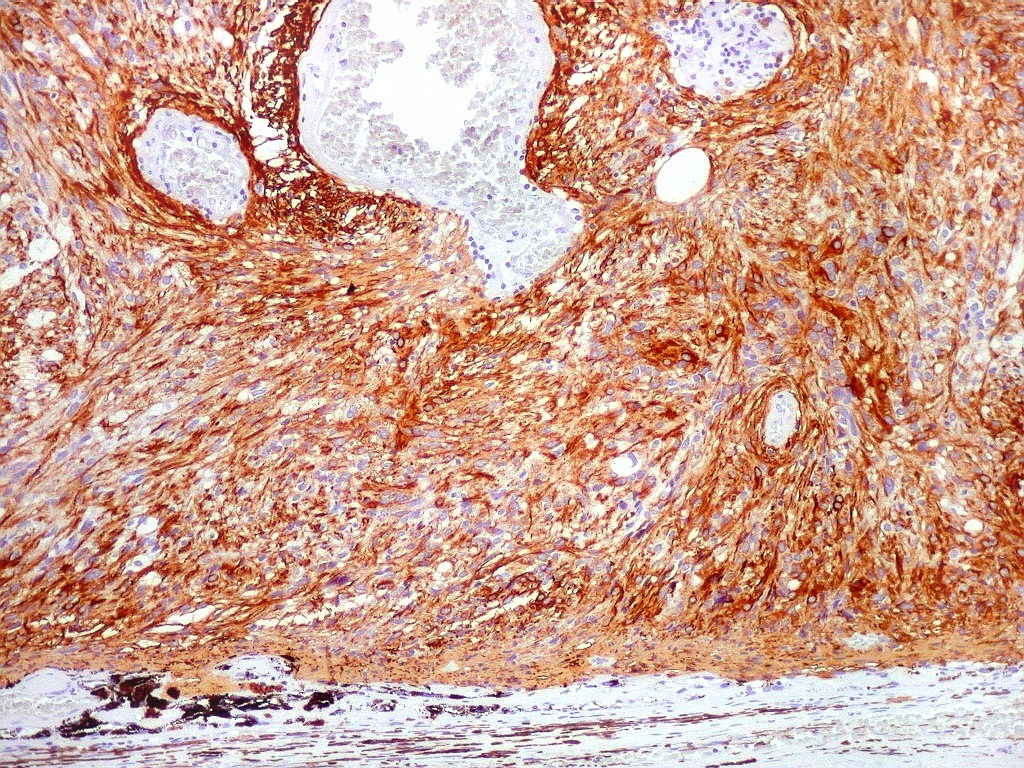 |
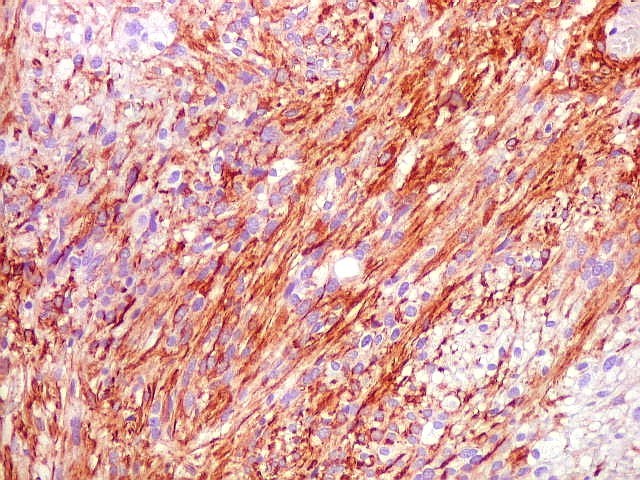
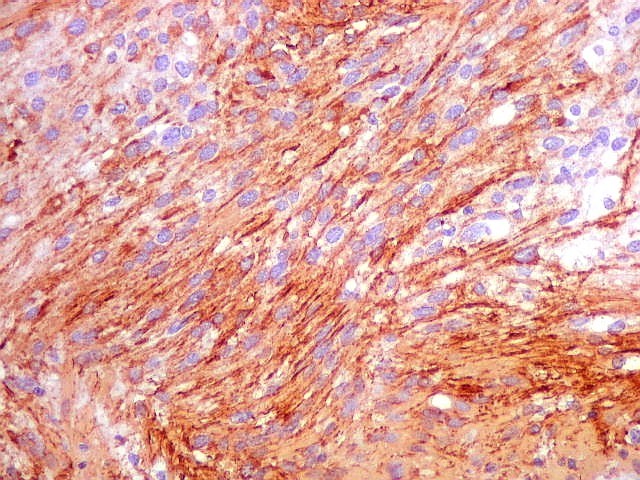
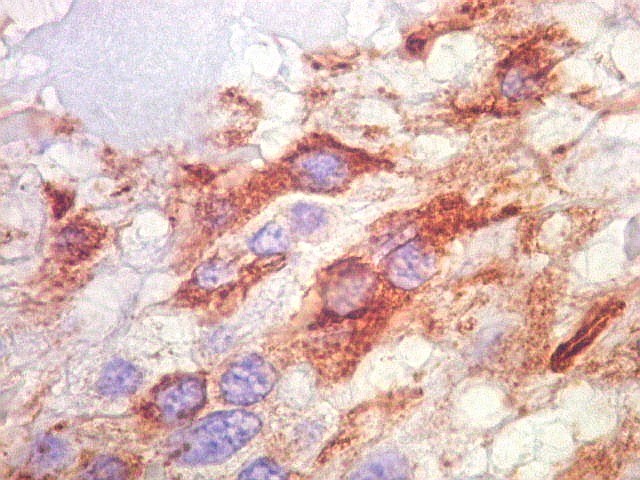
| GFAP. Astrócitos pilocíticos. Na maior parte do tumor reconhecem-se astrócitos de padrão morfológico pilocítico, alongados, bipolares, com núcleos ovalados, e prolongamentos delgados saindo dos polos da célula. Estes se arranjam em paralelo, lembrando uma cabeleira, o que origina o termo 'pilocítico'. Em outras áreas os feixes celulares são cortados obliqua- ou transversalmente. | |
 |
 |
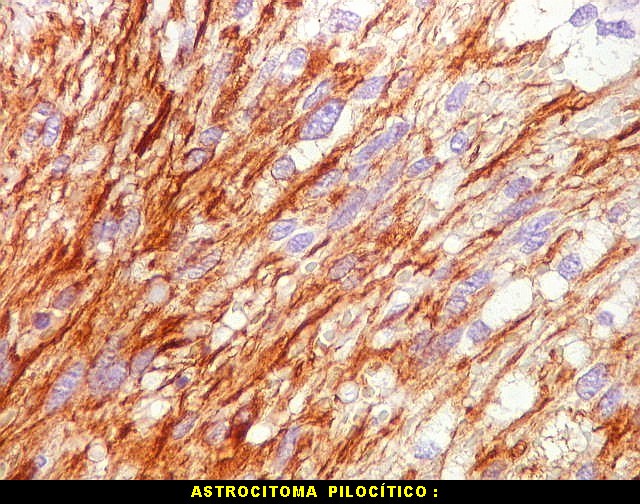 |
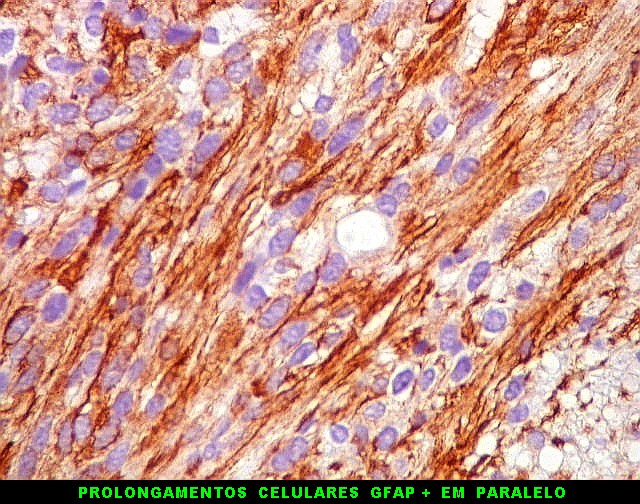 |
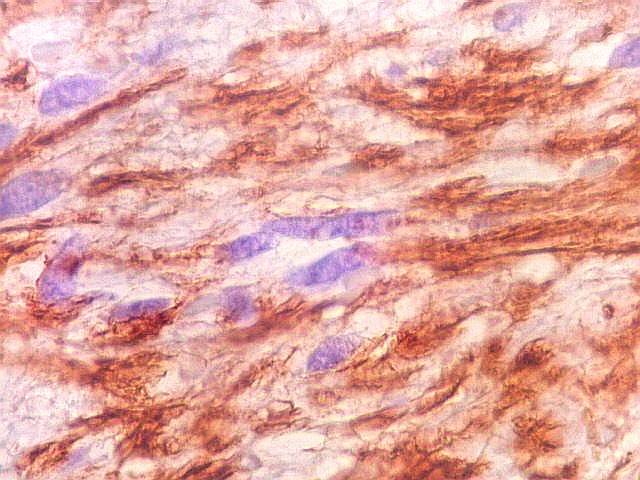 |
 |
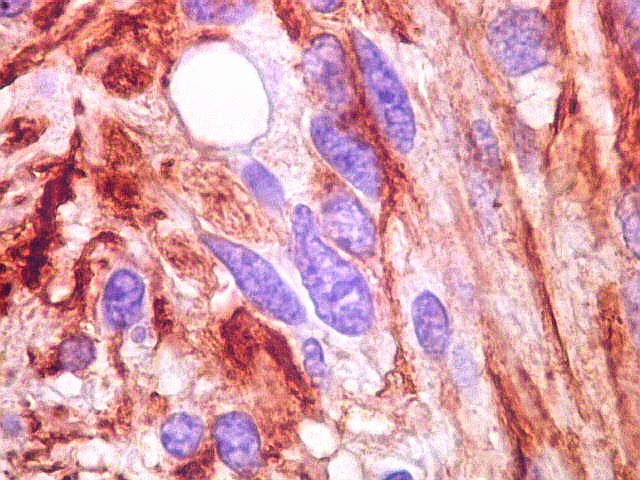 |
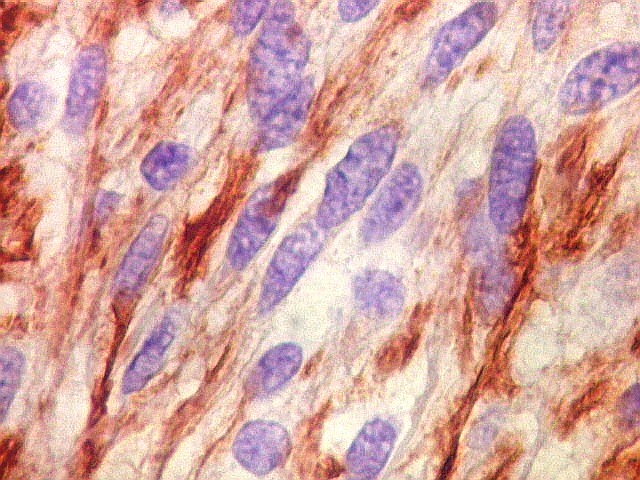 |
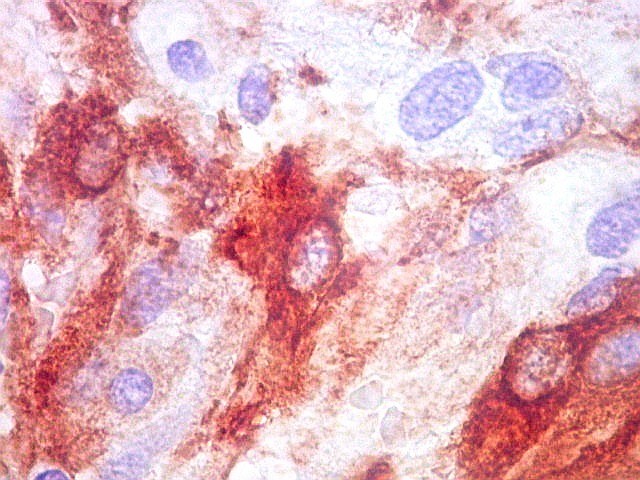
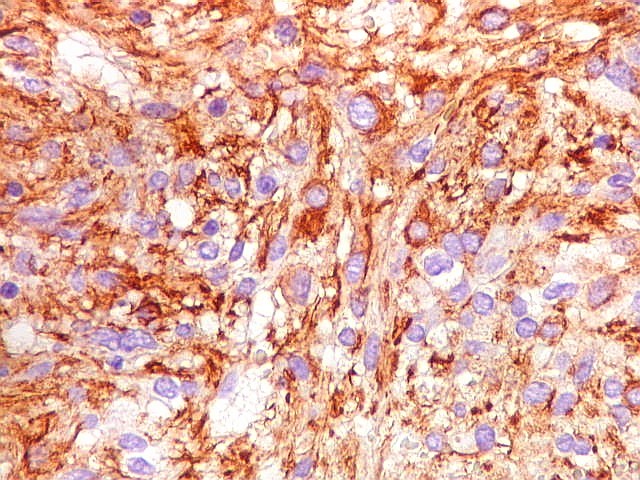
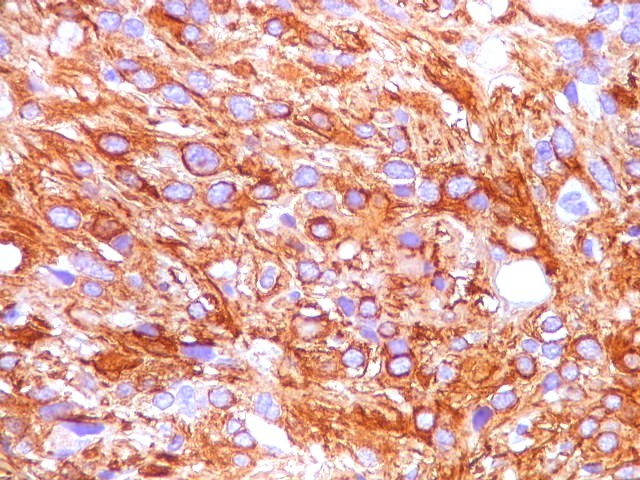
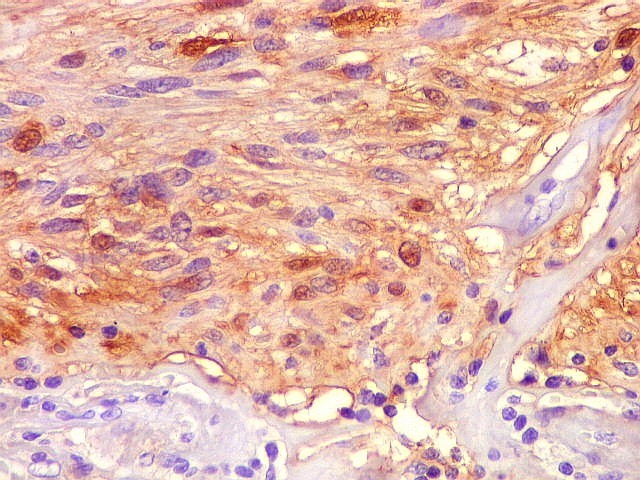
| GFAP. Feixes em cortes oblíquos. Variação de positividade entre células. É notável que células vizinhas possam ser positivas e negativas, mesmo lado a lado. | |
 |
 |
 |
 |
 |
 |
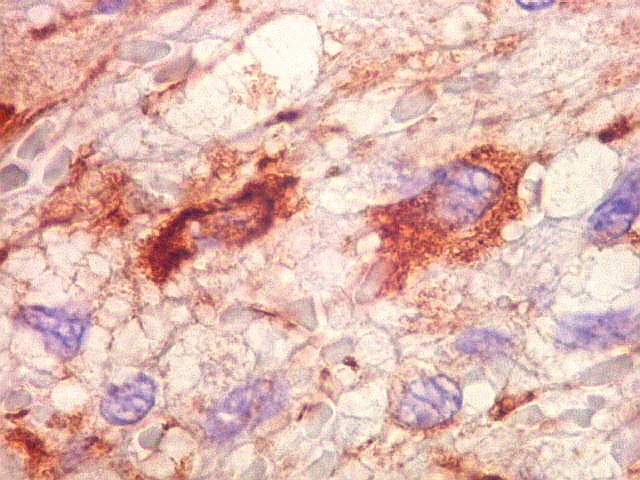
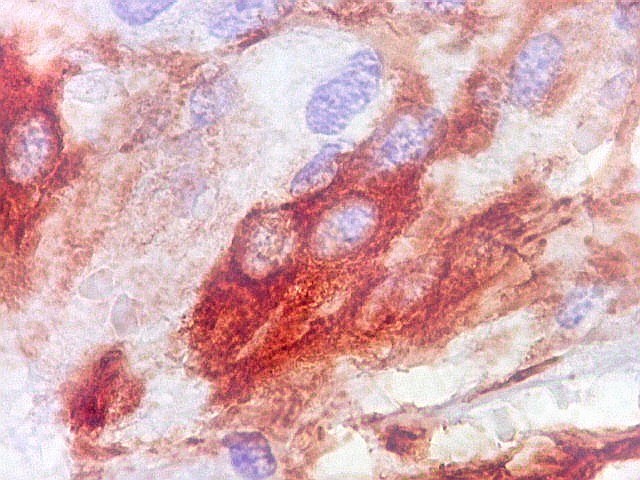
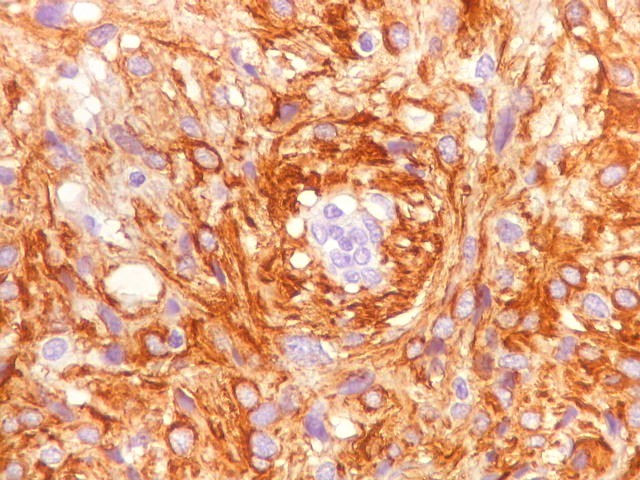
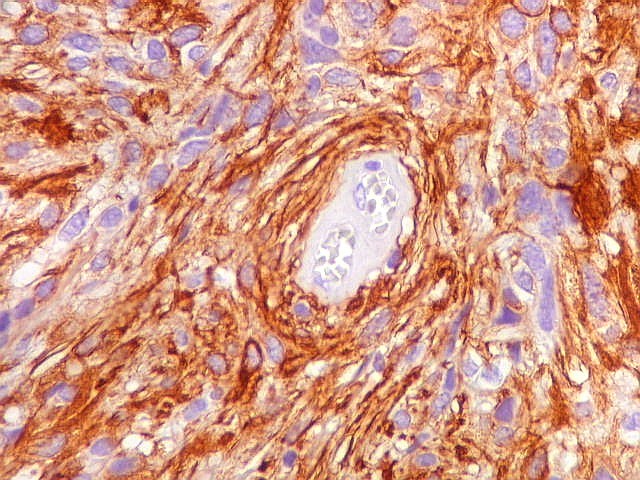
| GFAP. Vasos. São, como esperado, negativos para GFAP. Parece haver tendência a que as células mais próximas sejam mais fortemente positivas. | |
 |
 |
 |
|
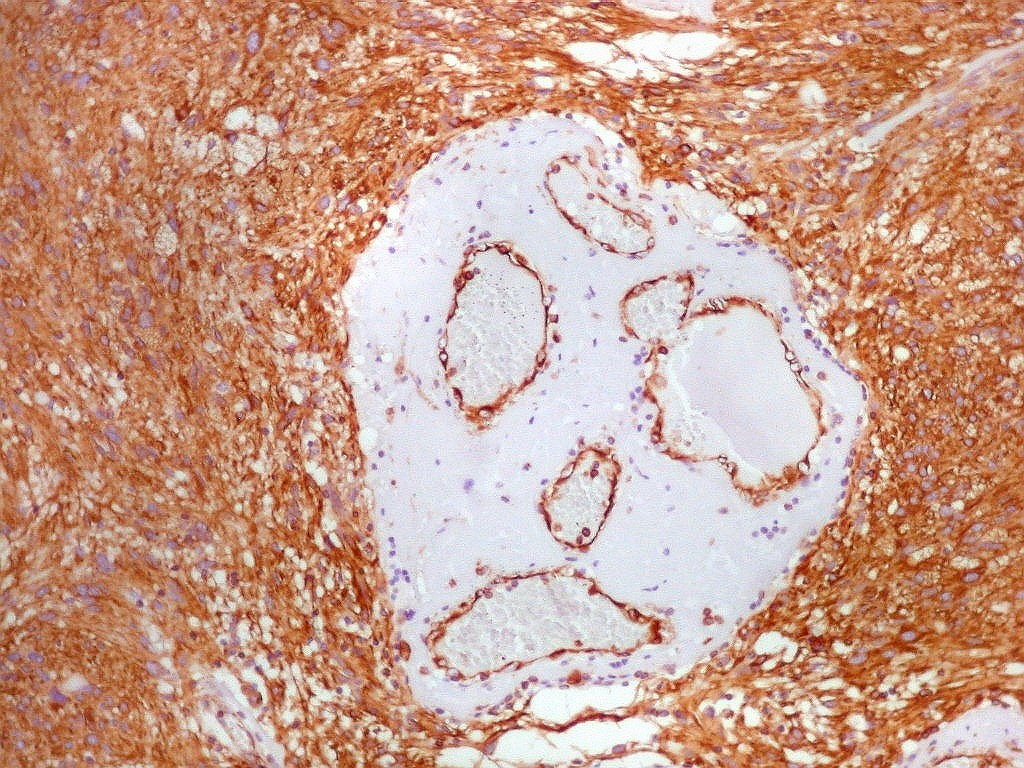
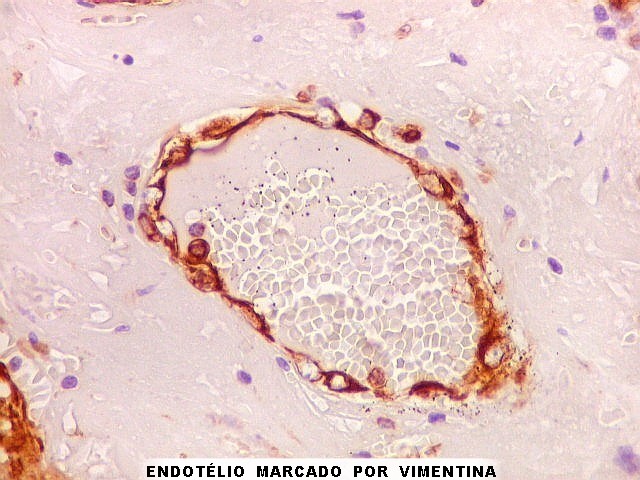
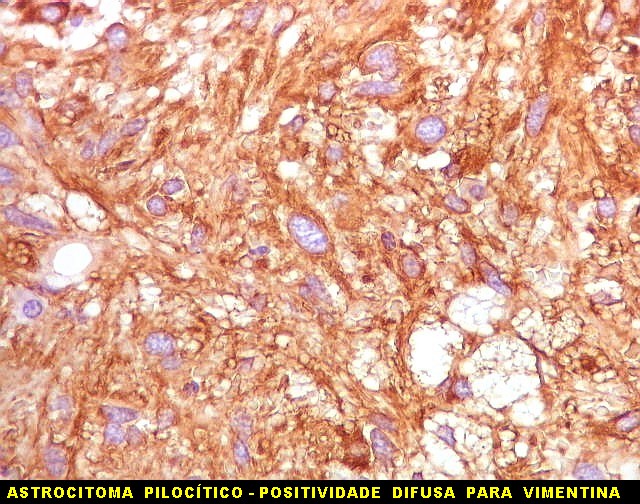
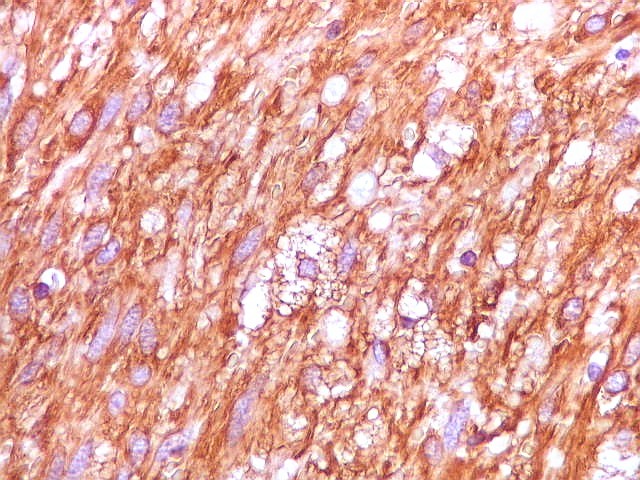
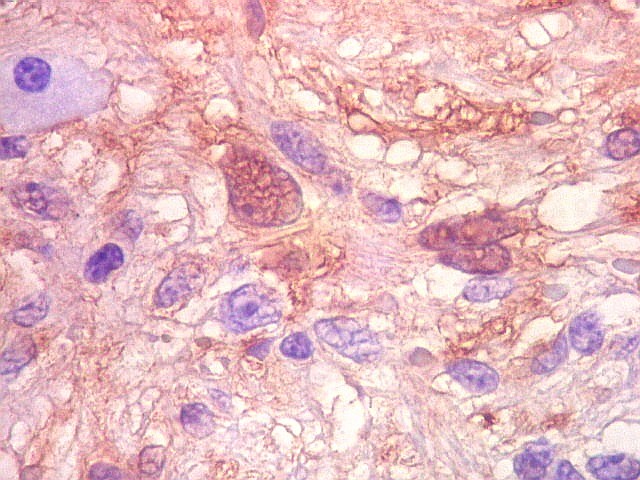
| Vimentina. As células neoplásicas são forte e difusamente positivas para vimentina, um filamento intermediário ubiquitário, mas expressado também em células de linhagem astrocitária. Ao contrário, GFAP só é positivo em astrócitos e células ependimárias. Aqui há marcação também do endotélio vascular, que fica realçado em relação ao tecido fibroso hialinizado e fibrina observados em muitos vasos do tumor. |
 |
|
 |
 |
 |
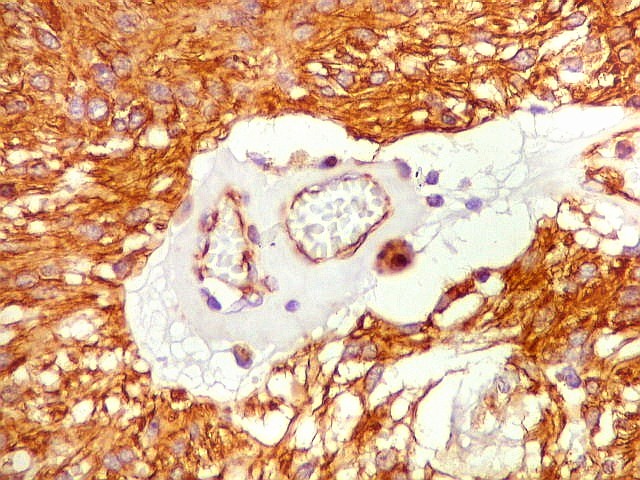
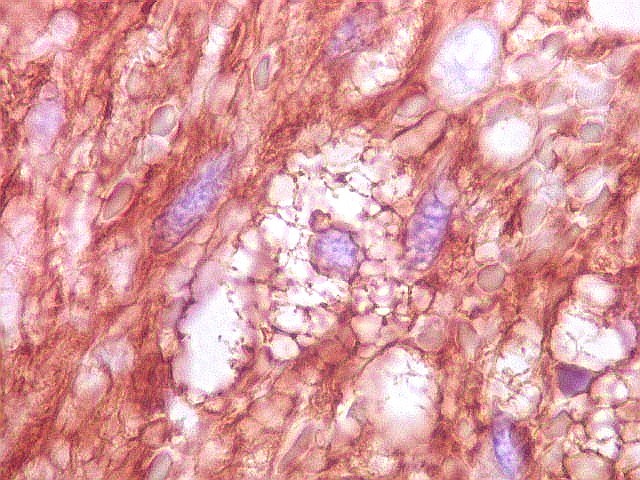
| Vimentina. Células vacuoladas. Algumas células de aspecto xantomatoso foram observadas em meio às neoplásicas, presumivelmente macrófagos. Não foram observadas com GFAP, assim não devem pertencer ao tumor. | |
 |
 |
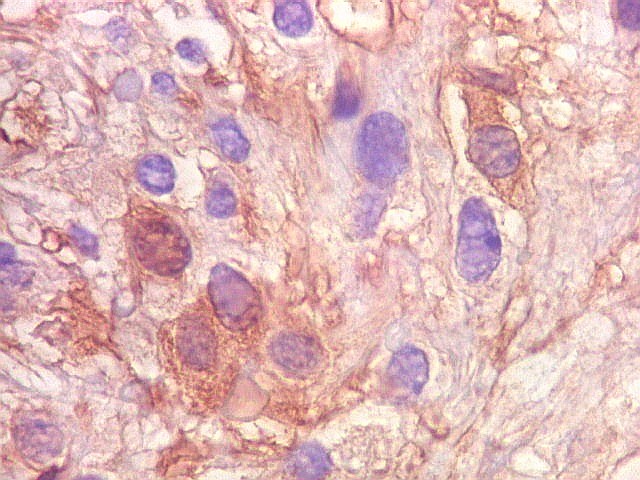
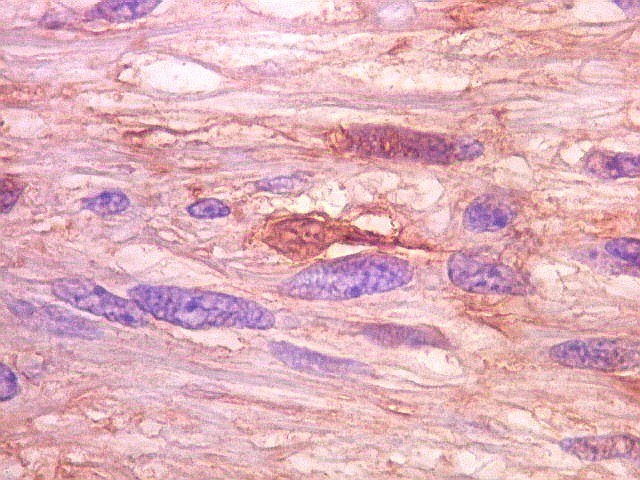
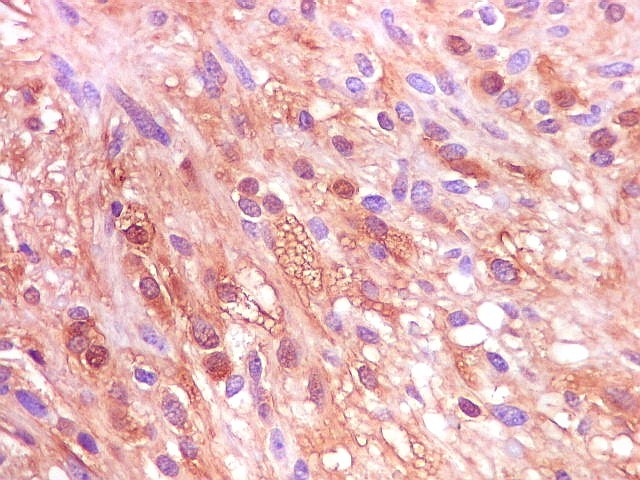
| S-100. Positividade para proteína S-100 (para breve texto clique) foi encontrada em parte das células do tumor, tanto nuclear como citoplasmática. Há notável variação na marcação, como notado para GFAP. | |
 |
 |
|
 |
 |
 |
 |
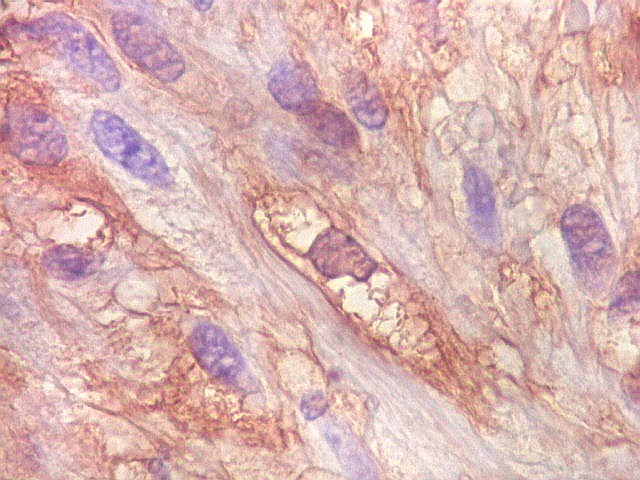 |
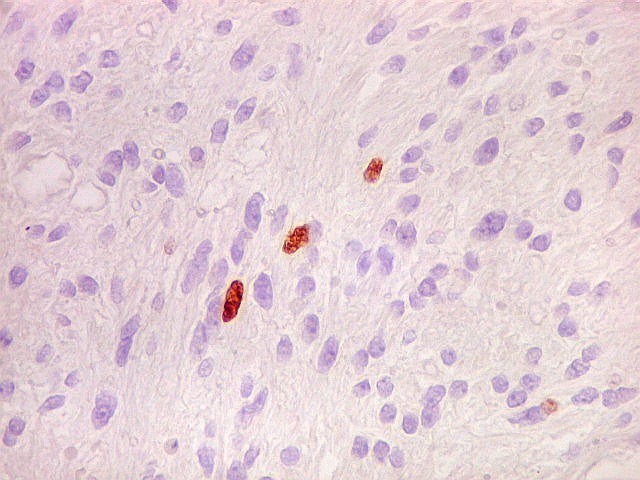
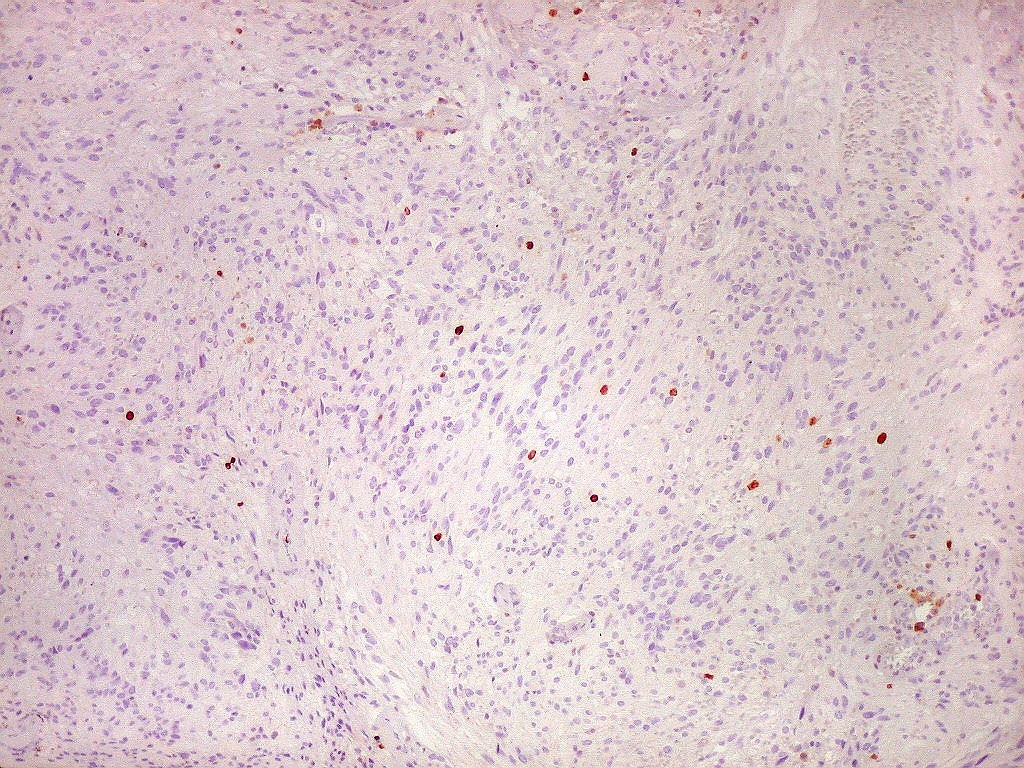
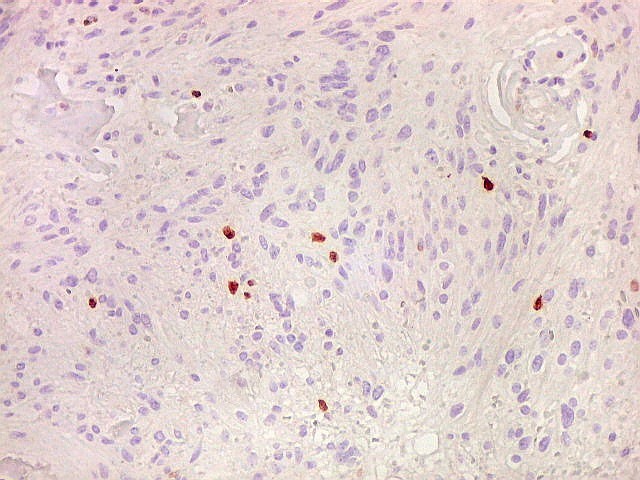
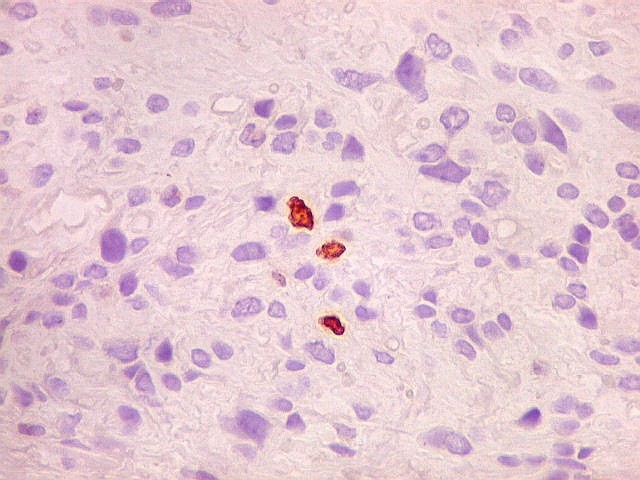
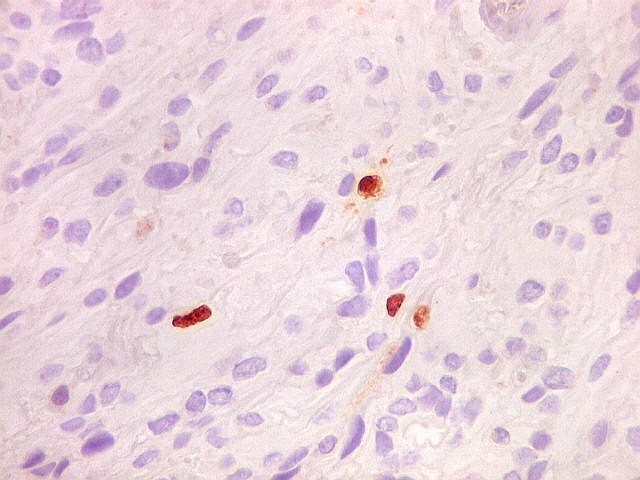
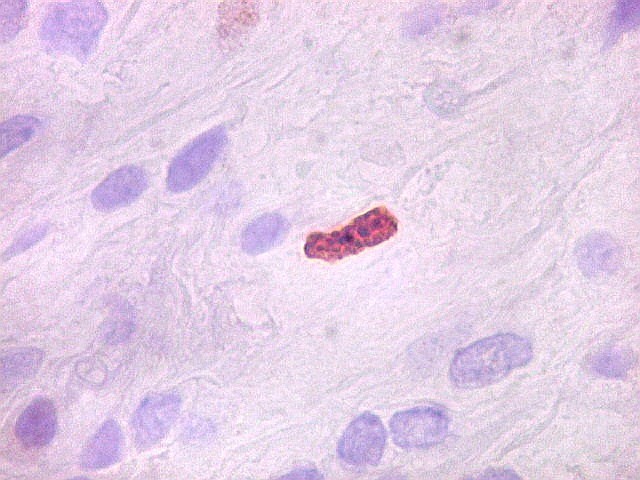
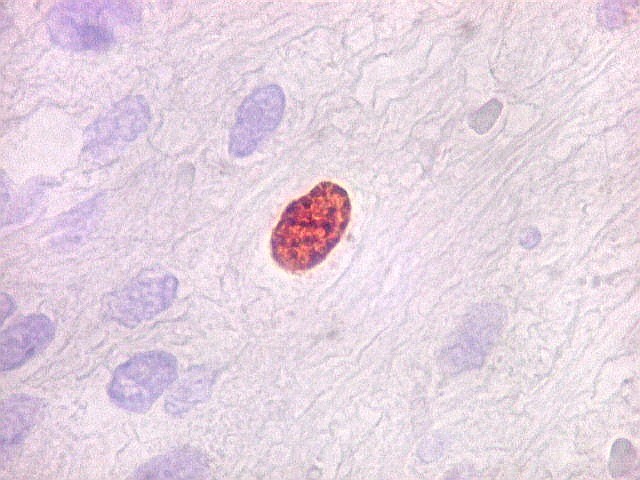
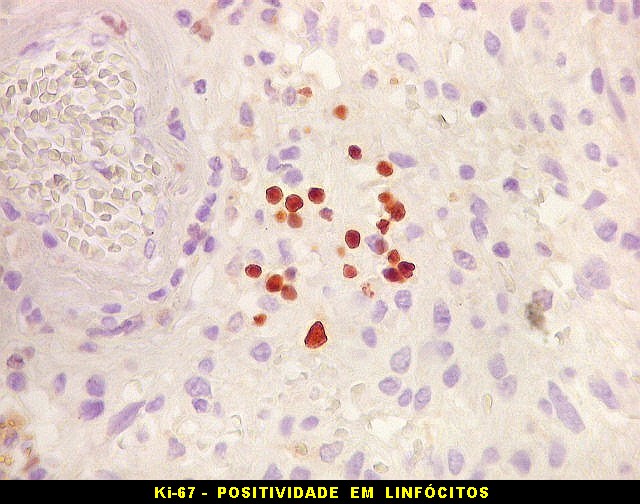
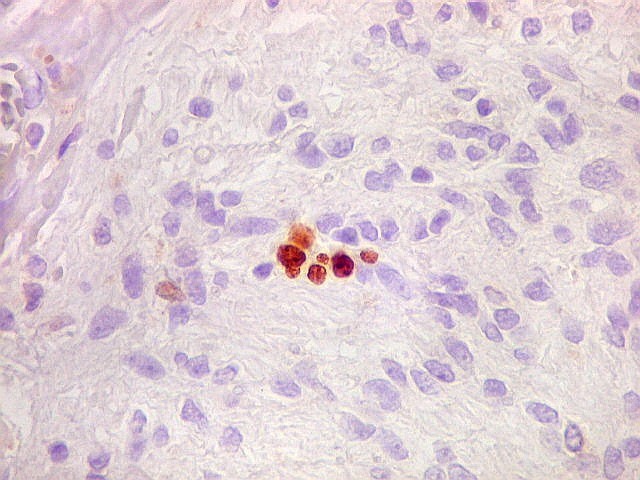
| Ki-67. Houve positividade de um número modesto de células (da ordem de 1 a 3%) para o marcador de proliferação celular Ki-67, indicando tumor de lento crescimento. Algumas células hematológicas (presumivelmente linfócitos) marcavam-se em proporção alta, o que é compatível com a normalidade (fotos da última fileira). |
 |
 |
|
 |
 |
 |
 |
 |
 |
| Astrocitoma
intraocular.
Astrocitomas originados no interior do olho podem ter dois padrões histológicos. Um deles caracteriza-se por feixes entrelaçados de células fusiformes com uma minoria de células poligonais. O outro é constituído por astrócitos gemistocíticos, com células grandes de citoplasma abundante em vidro fosco, indistinguíveis das do astrocitoma subependimário de células gigantes da esclerose tuberosa. Ambos tipos expressam GFAP difusamente, são biologicamente benignos e associados a síndromes disgênicas, particularmente à esclerose tuberosa. Tumores com gemistócitos mostram maior tendência a crescimento invasivo. Definição. O termo astrocitoma intraocular designa uma neoplasia de baixo grau histológico, mas com crescimento progressivo e autônomo, que se origina na retina ou no nervo óptico anteriormente à lamina cribrosa sclerae (ou cabeça do nervo óptico). Embora histologicamente benignos, podem resultar em morbidade ocular e perda da visão. Epidemiologia. O tumor é raro, pode ser assintomático, e o diagnóstico só em bases clínicas fica presuntivo (o diagnóstico definitivo é histológico). Isto contribui para a falta de estimativas populacionais sobre sua prevalência. Numa revisão de 42 astrocitomas intraoculares confirmados histologicamente, 57% estavam associados à esclerose tuberosa, 14% à neurofibromatose do tipo 1 e 29% foram esporádicos. O complexo da esclerose tuberosa, cuja prevalência estimada é cerca de 1:10.000, é a doença hereditária mais comumente associada com astrocitomas intraoculares. Cerca de metade dos pacientes de esclerose tuberosa têm astrocitomas da retina ou nervo óptico, na maioria lesões clinicamente estacionárias classificadas como hamartomas. Clínica. Na maioria, os globos oculares em que astrocitoma foi histologicamente confirmado foram removidos devido à amaurose, dor ocular ou por simular retinoblastoma em crianças ou melanoma amelanótico ou carcinoma metastático em adultos. A idade variou de 1 mês a 45 anos, e o tamanho desde alguns milímetros a massas preenchendo o globo. Pelo menos metade tem calcificações. Em comparação com astrocitomas, os hamartomas astrocitários de nervo óptico ou retina apresentam-se como massas bem delimitadas, esbranquiçadas ou amareladas na oftalmoscopia, com aparência gelatinosa ou translúcida, variando de < 1 mm a > 5 mm. Ficam estáveis ou com mínimo crescimento. Em pacientes com esclerose tuberosa, podem ser monitorados com fotografia e angiografia com fluoresceína por longos períodos sem necessidade de outras intervenções. Astrocitose (gliose) reacional da retina é histologicamente semelhante ao astrocitoma, mas clinicamente difere por ocorrer geralmente em olhos já afetados por outra condição primária como retinopatia da prematuridade, ou retinopatia diabética proliferativa. Histologicamente, o astrocitoma de padrão pilocítico mostra células alongadas com núcleos ovalados, e citoplasma róseo mal delimitado, arranjadas em feixes multidirecionados. Não se observam mitoses ou necrose. Depósitos de cálcio são comuns. Fibras de Rosenthal e corpos hialinos não haviam sido documentados (mas ver caso acima). Não há diferenças histopatológicas entre tumores esporádicos ou sindrômicos. O outro padrão de astrocitoma intraocular assemelha-se ao astrocitoma subependimário de células gigantes da esclerose tuberosa. As células têm aspecto de astrócitos gemistocíticos, com núcleos arredondados, por vezes com nucléolos proeminentes, citoplasma abundante e róseo, em vidro fosco, e abundantes calcoesferitos (concreções calcáreas). Figuras de mitose são raras ou ausentes, mas necrose é comum e pode atingir 50% do tumor. Há relatos de invasão da coróide e esclera, e extensão atrás da lamina sclerae do nervo óptico. Em ambos padrões há positividade difusa para GFAP e S-100. Nos astrocitomas de células gigantes pode haver co-expressão de antígenos neuronais como a enolase neurônio-específica (NSE). Patogênese. Esclerose tuberosa ou doença de Bourneville. Uma proporção substancial dos astrocitomas e hamartomas astrocitários intraoculares está associada à esclerose tuberosa, uma doença sistêmica de fenótipo único relacionada a mutações em 2 genes distintos (TSC1 e TSC2). A doença é causada por perda de função de um dos genes, que leva à perda de uma das respectivas proteínas, hamartina e tuberina. Ambas se ligam formando um complexo que regula a via de transdução P13K, influenciando o crescimento e sobrevida celulares. Neurofibromatose do tipo 1 ou de von Recklinghausen é menos freqüentemente associada a astrocitomas intraoculares que a esclerose tuberosa, mas é a síndrome genética que mais comumente causa astrocitomas das vias ópticas anteriores, como o glioma de nervo óptico, que é histologicamente um astrocitoma pilocítico. As alterações moleculares em astrocitomas variam com a localização. Astrocitomas pilocíticos associados à NF1 são devidos à inativação homozigótica do gene NF1. Já tumores esporádicos com a mesma morfologia no cerebelo ou no nervo óptico podem ocorrer independentemente da mutação. As mutações (se houver) no astrocitoma esporádico da retina não são conhecidas. Fonte. Pusateri A; Margo CE, Intraocular astrocytoma and its differential diagnosis. Arch Pathol Lab Med. 2014; 138: 12501254. Outras referências.
|
| Agradecimentos. Caso estudado com o residente de segundo ano (R2) Dr. Gabriel Ledo Pereira de Oliveira. Processamento histológico e lâminas HE pelo pessoal do Laboratório de Rotina: Mariagina de Jesus Gonçalves, Maria José Tibúrcio, Guaracy da Silva Ribeiro, Fernando Wagner dos Santos Cardoso, Viviane Ubiali, Fernanda das Chagas Riul e Vanessa Natielle Pereira de Oliveira. Procedimentos imunohistoquímicos pelo pessoal do Laboratório de Pesquisa - Ana Claudia Sparapani Piaza, Luzia Aparecida Magalhães Ribeiro Reis e Arethusa de Souza. Depto de Anatomia Patológica da FCM-UNICAMP, Campinas, SP. |
| Para TC, RM desta paciente, clique » |  |
| Texto - astrocitoma intraocular | |
| Mais sobre astrocitomas pilocíticos: |
| Na graduação | Texto ilustrado | Casos, neuroimagem | Casos, neuropatologia | Características de imagem | Glioma de nervo óptico | Microscopia eletrônica (1) (2) |
| Neuropatologia
- Graduação |
Neuropatologia -
Estudos de casos |
Neuroimagem
- Graduação |
Neuroimagem -
Estudos de Casos |
Roteiro
de aulas |
Textos
de apoio |
Correlação
Neuropatologia - Neuroimagem |
| Índice alfabético - Neuro | Adições recentes | Banco de imagens - Neuro | Textos ilustrados | Neuromuscular | Patologia - outros aparelhos | Pages in English |
|
|