|
|
1. Esfregaço, parafina - HE. |
|
|
|
1. Esfregaço, parafina - HE. |
|
| Masc. 28 a. Clique para : história clínica, TC, RM, imunohistoquímica para marcadores gliais, neuronais, outros. |
| Destaques da microscopia. Textos sobre astrocitoma subependimário de células gigantes, esclerose tuberosa e comentário sobre o presente caso. | ||
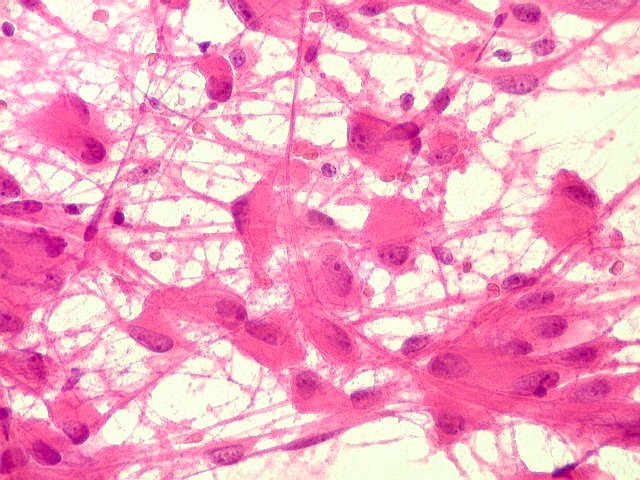
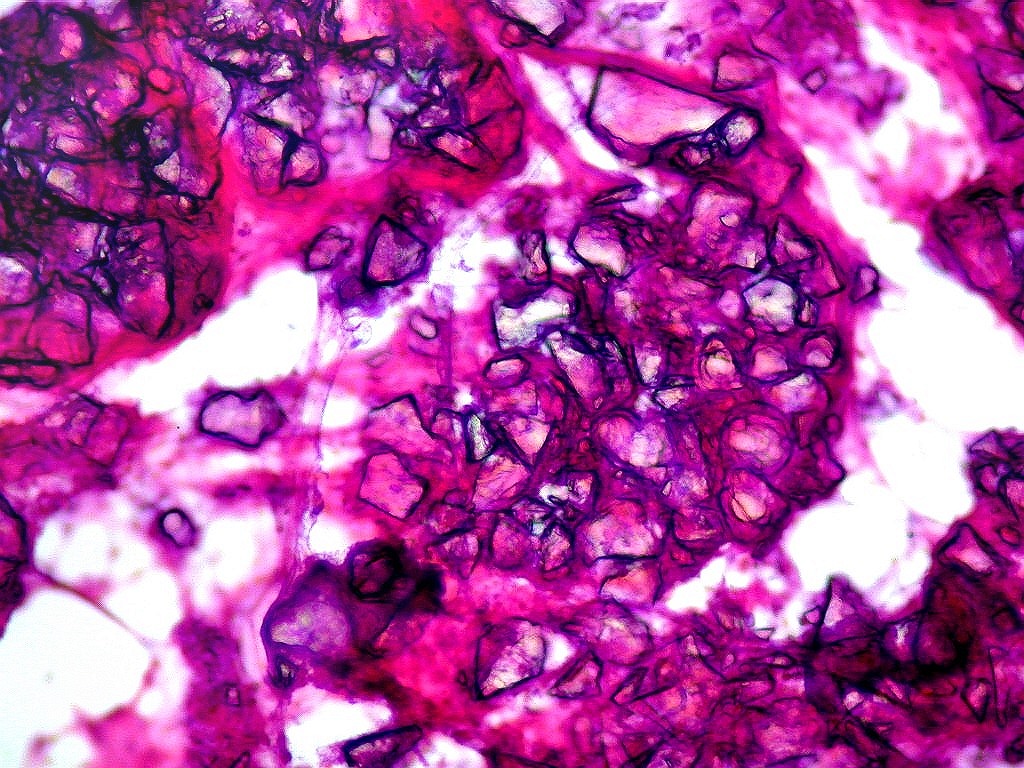
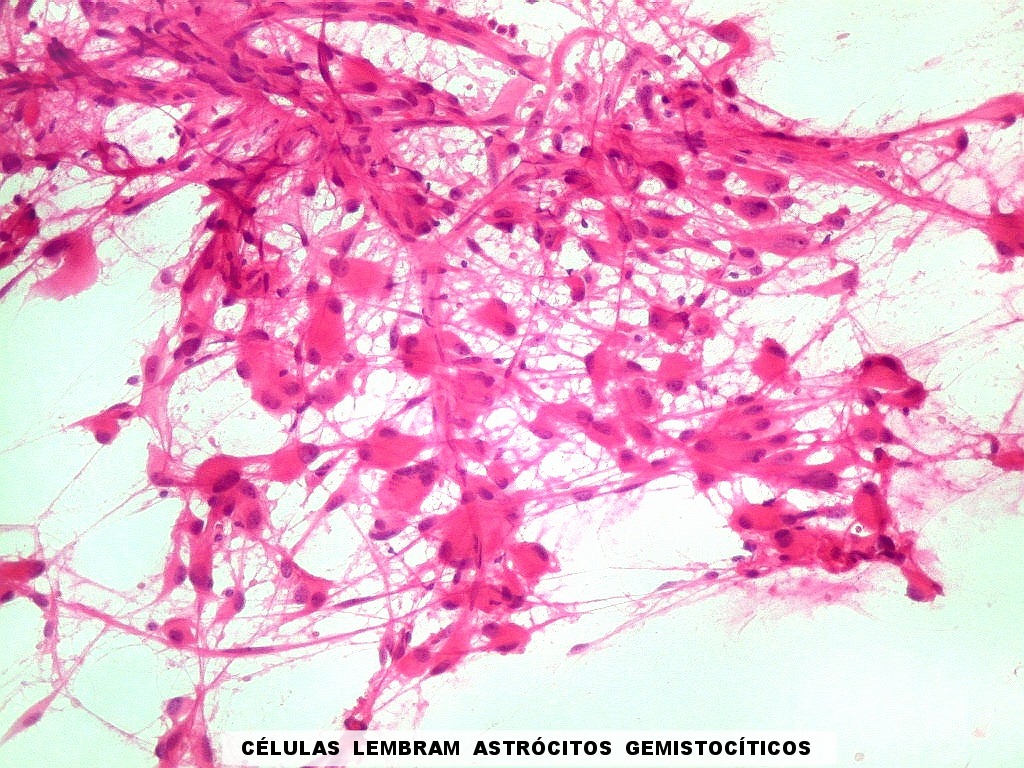
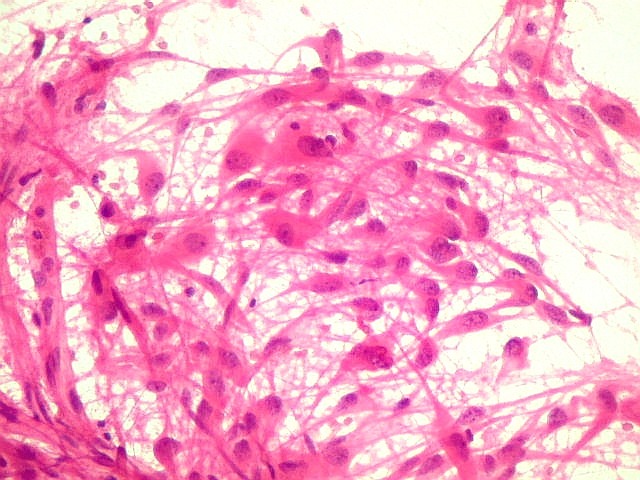
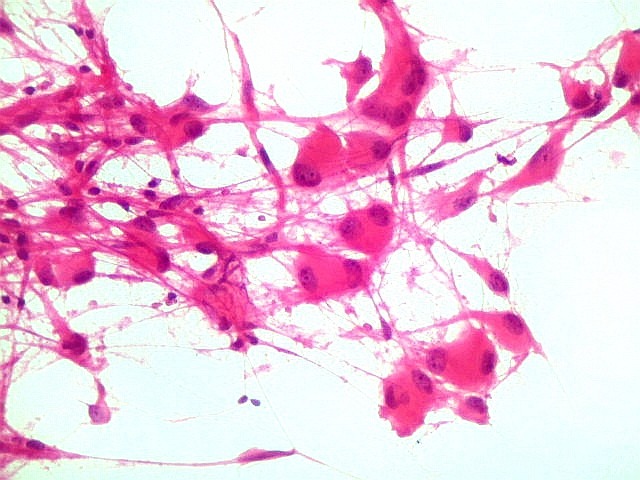
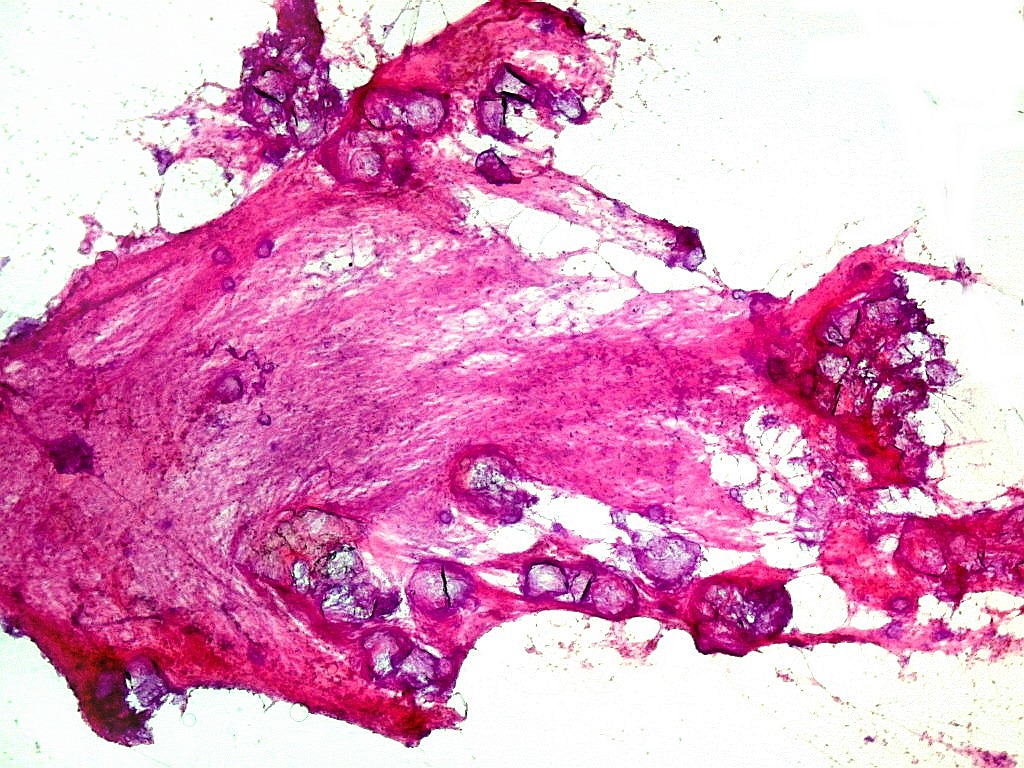
| Esfregaço. Células lembrando astrócitos gemistocíticos | Calcificações. | Parafina HE. Células polimórficas, atipias nucleares, por vezes disposição em feixes. |
 |
 |
 |
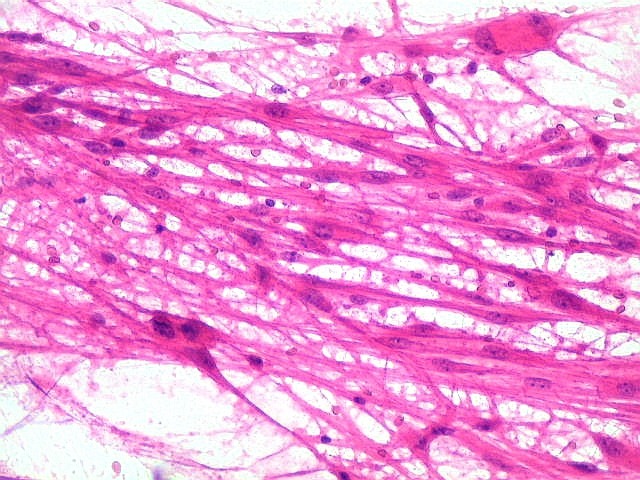
| Células volumosas, núcleo excêntrico, lembrando astrócitos gemistocíticos. | Idem, com nucléolos lembrando neurônios. | Área pilocítica com fibras de Rosenthal. |
 |
 |
 |
| Mitoses, não raras, geralmente típicas | Agrupamentos de macrófagos xantomatosos. | Calcificações |
 |
 |
 |
| Para destaques da imunohistoquímica, clique. | ||
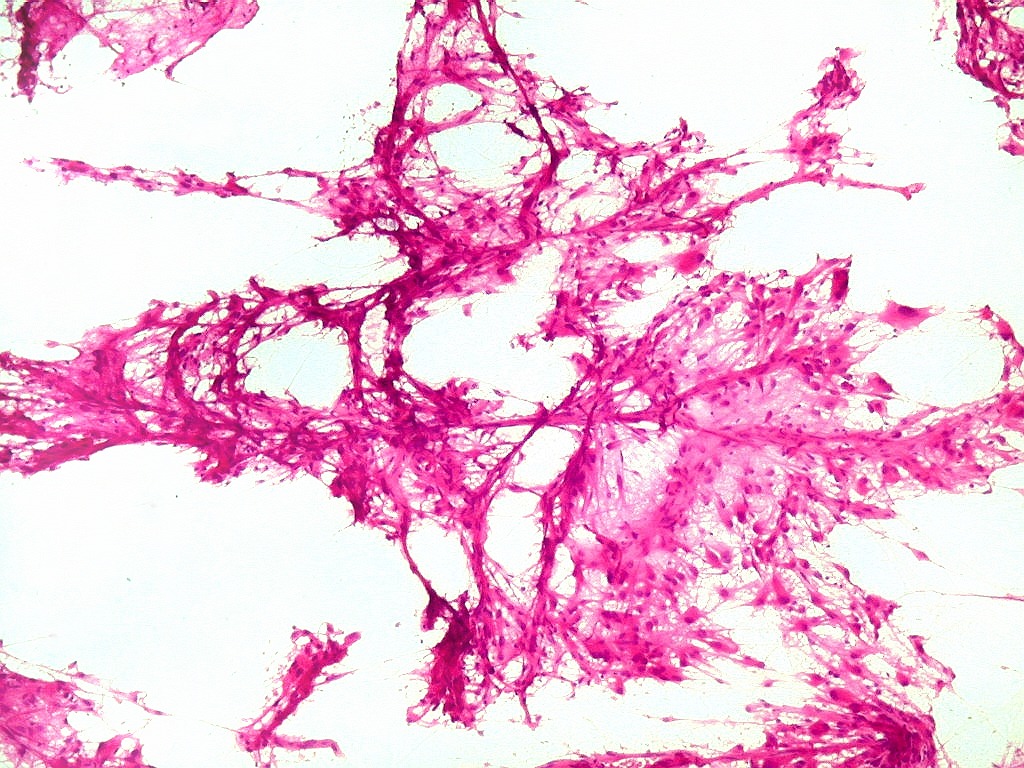
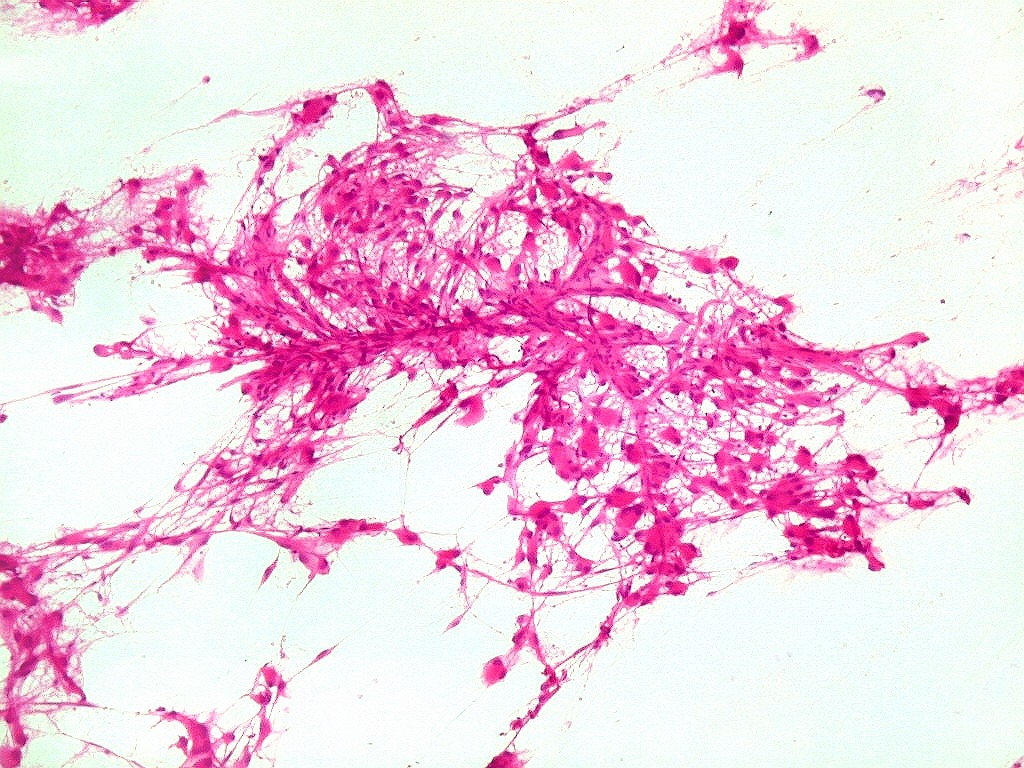
| ESFREGAÇO. Pequenos fragmentos de tecido a fresco, obtidos durante o ato cirúrgico, são esfregados por deslizamento entre duas lâminas, fixados rapidamente em álcool absoluto, corados por HE, desidratados e montados em resina, como para cortes de parafina. As lâminas devem estar apoiadas sobre uma superfície firme (a bancada) e a pressão aplicada na lâmina de cima deve permitir espalhar o tecido na maior extensão possível da lâmina de baixo (a que vai ser corada). Pressão demais causa artefatos de esmagamento e danifica as células. Pressão de menos leva a espessura excessiva do preparado. Tecidos moles, como tumores neuroectodérmicos, se prestam bem. Tecidos fibrosos ou elásticos (como tumores conjuntivos ou cartilaginosos) não permitem bom espalhamento do tecido. Quando bem sucedida, a técnica é de grande valia, por extrair informações importantes de um mínimo de material (1 mm3), com rapidez (2 min. do recebimento do material à lâmina pronta). |
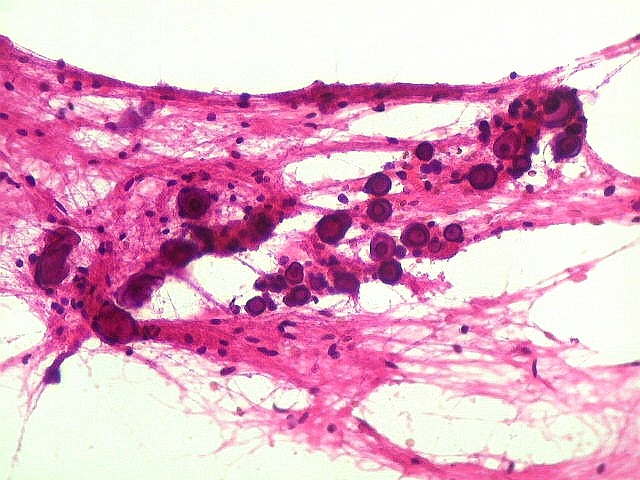
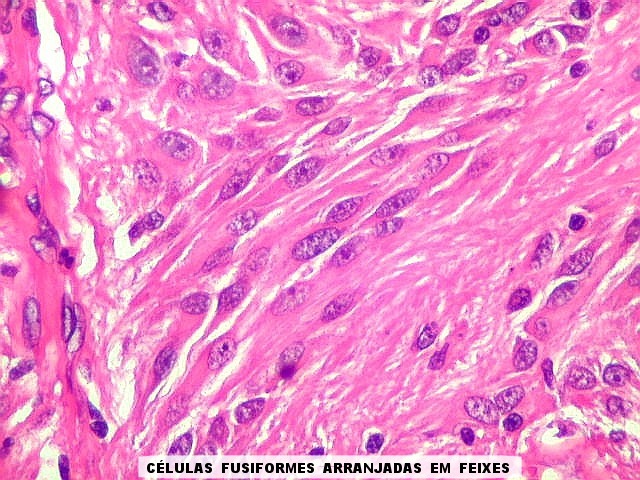
| Esfregaço.
O tumor
é constituído por células de citoplasma abundante,
associadas a rica rede vascular. Os vasos são finos e amplamente
ramificados. As células na maioria lembram astrócitos gemistocíticos.
Em outras áreas são alongadas e dispostas em feixes.
|
 |
 |
 |
|
 |
 |
 |
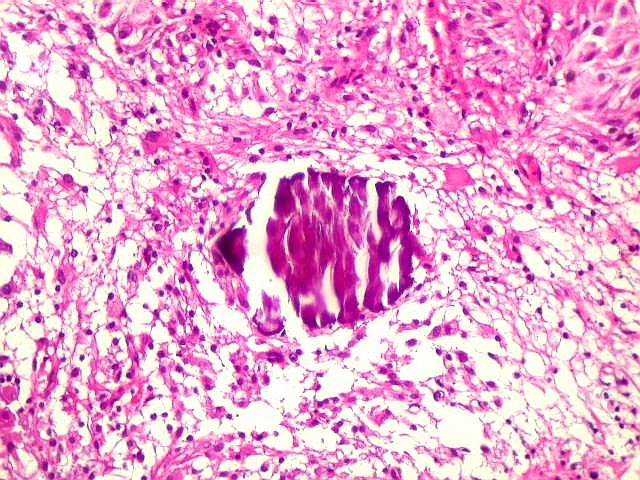
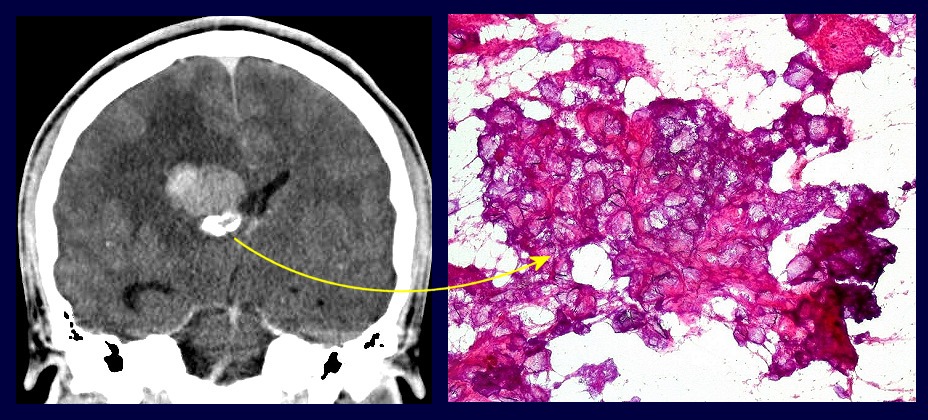
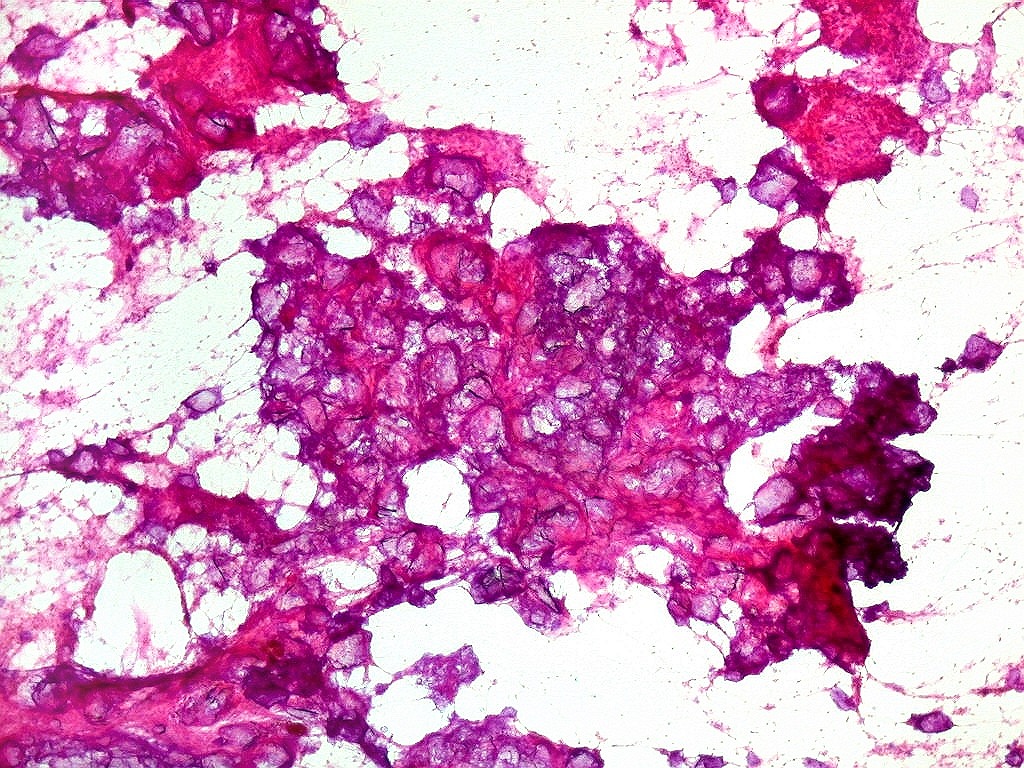
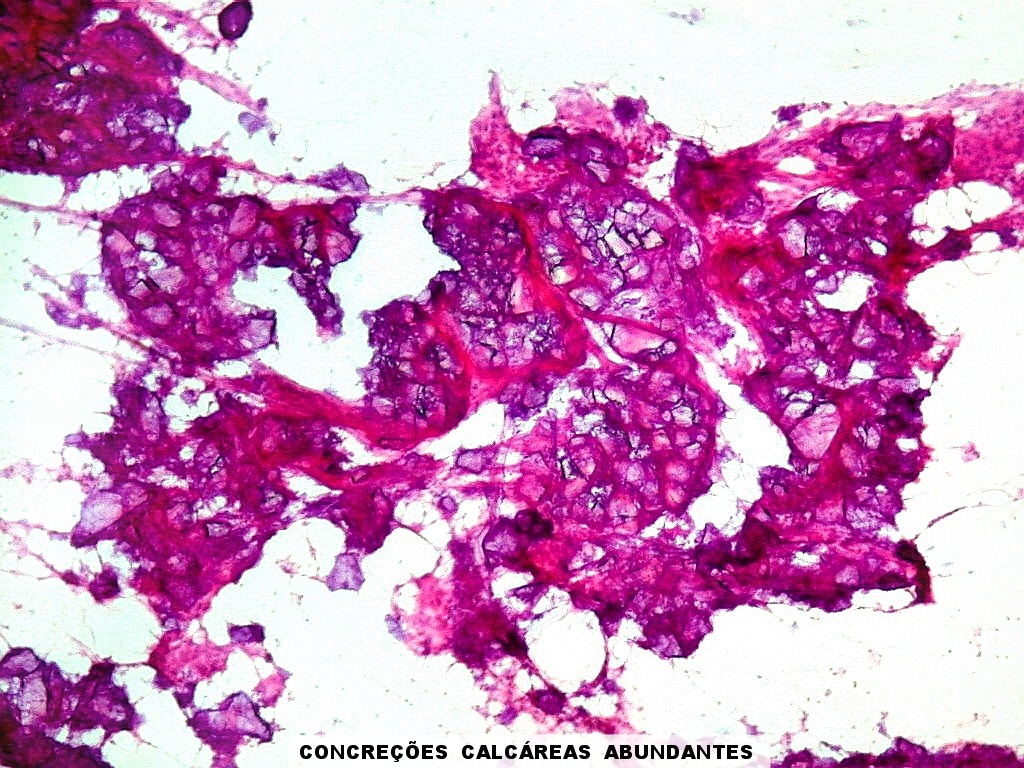
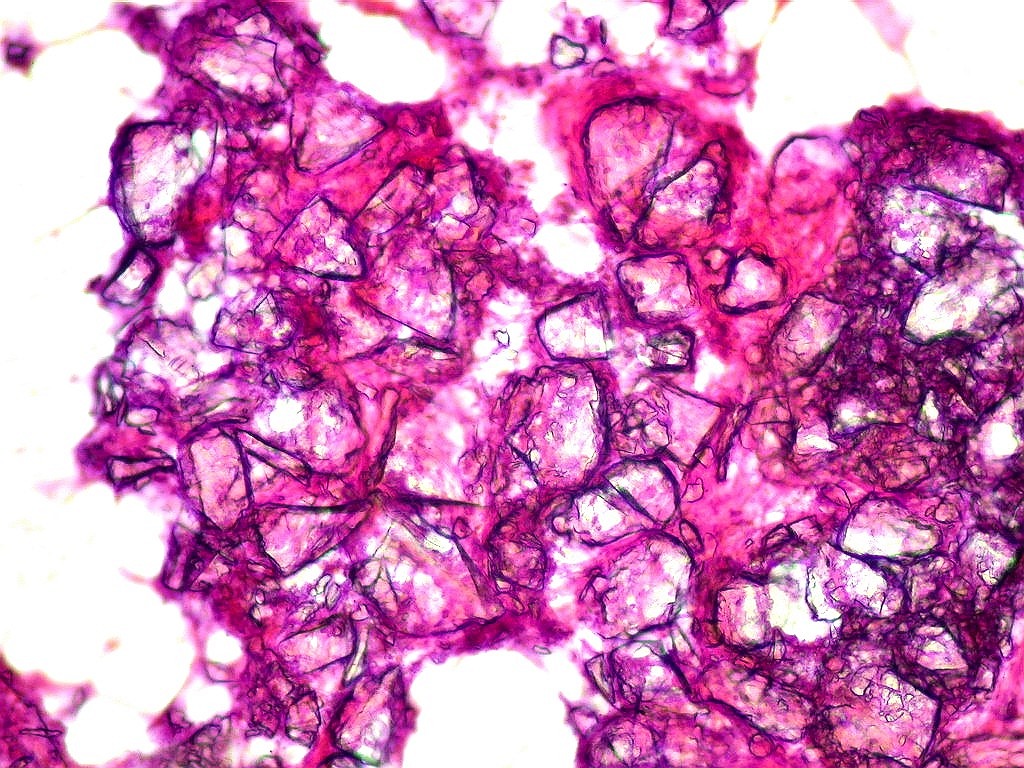
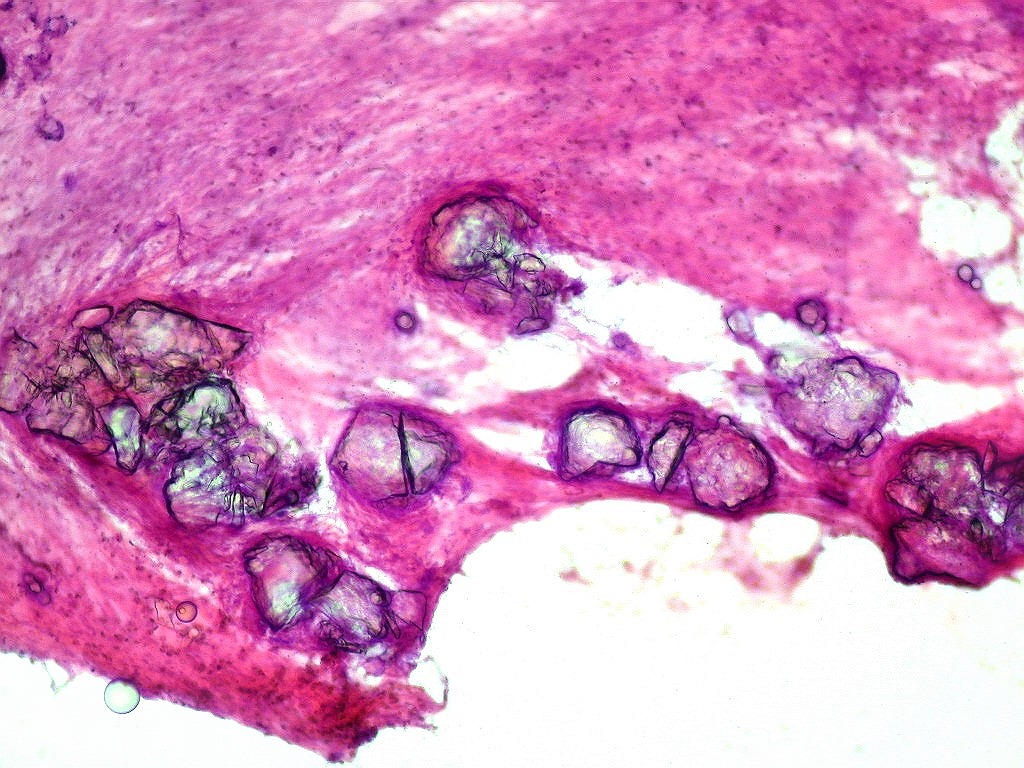
| Calcificações.
Alguns fragmentos abrigavam grande quantidade de concreções calcáreas. Provavelmente correspondiam a material retirado próximo à base de implantação do tumor no ventrículo lateral, onde calcificações grosseiras eram notadas na TC. Para TC e RM clique. |
 |
 |
 |
 |
 |
 |
 |
 |
|
|
|
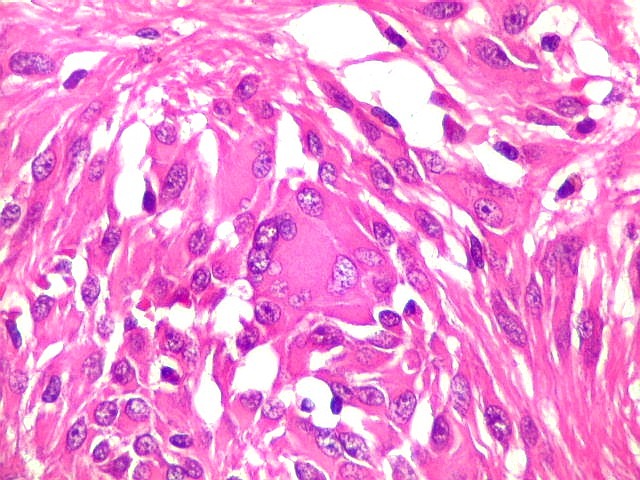
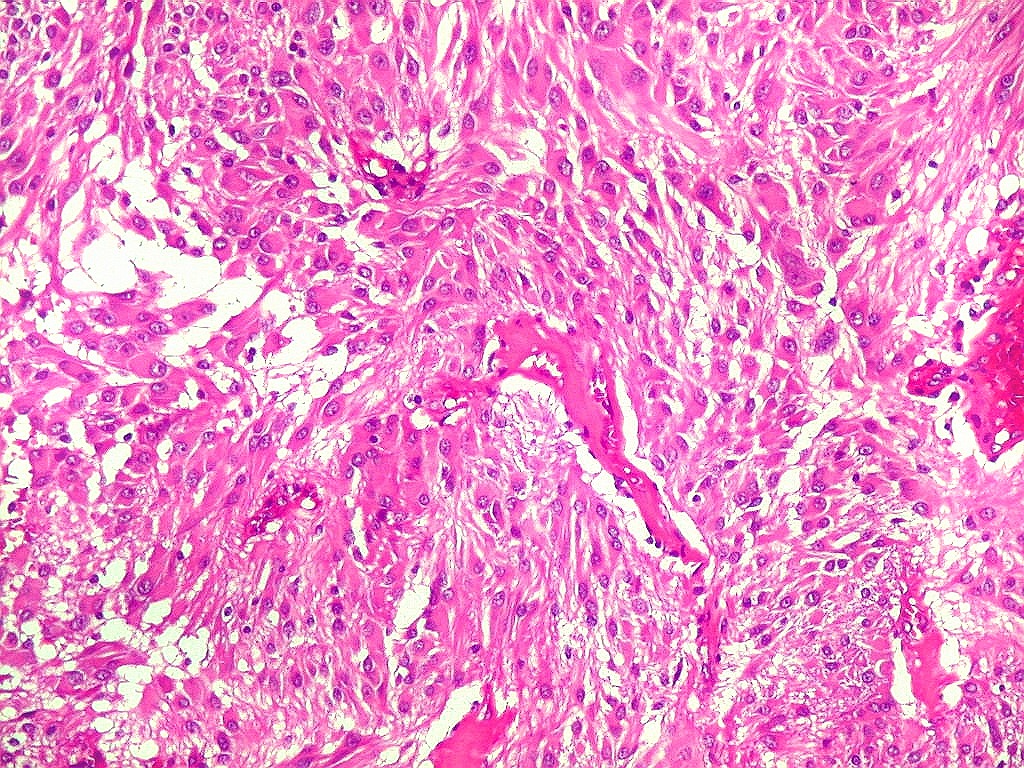
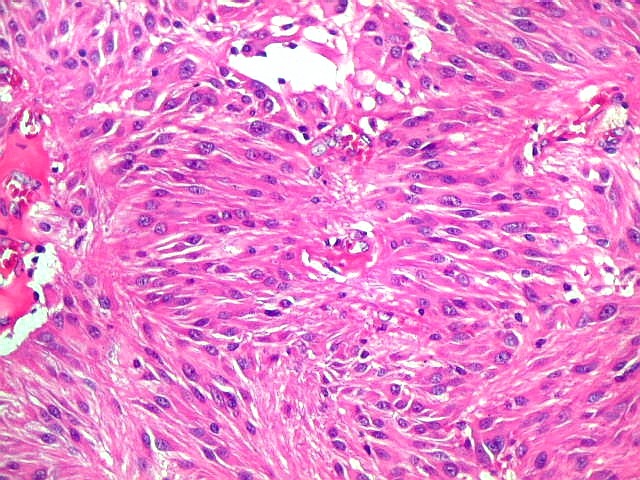
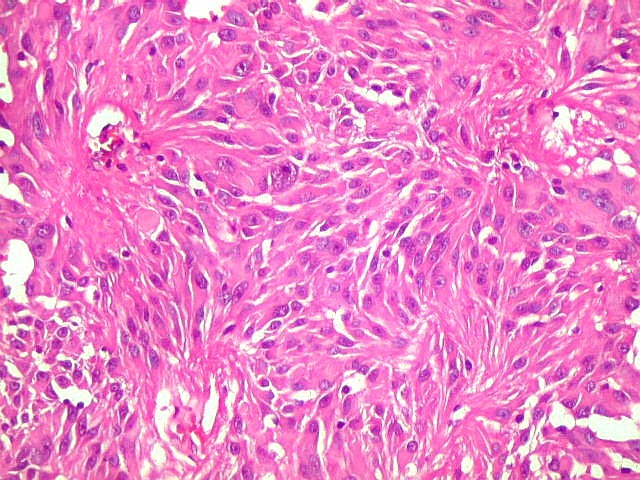
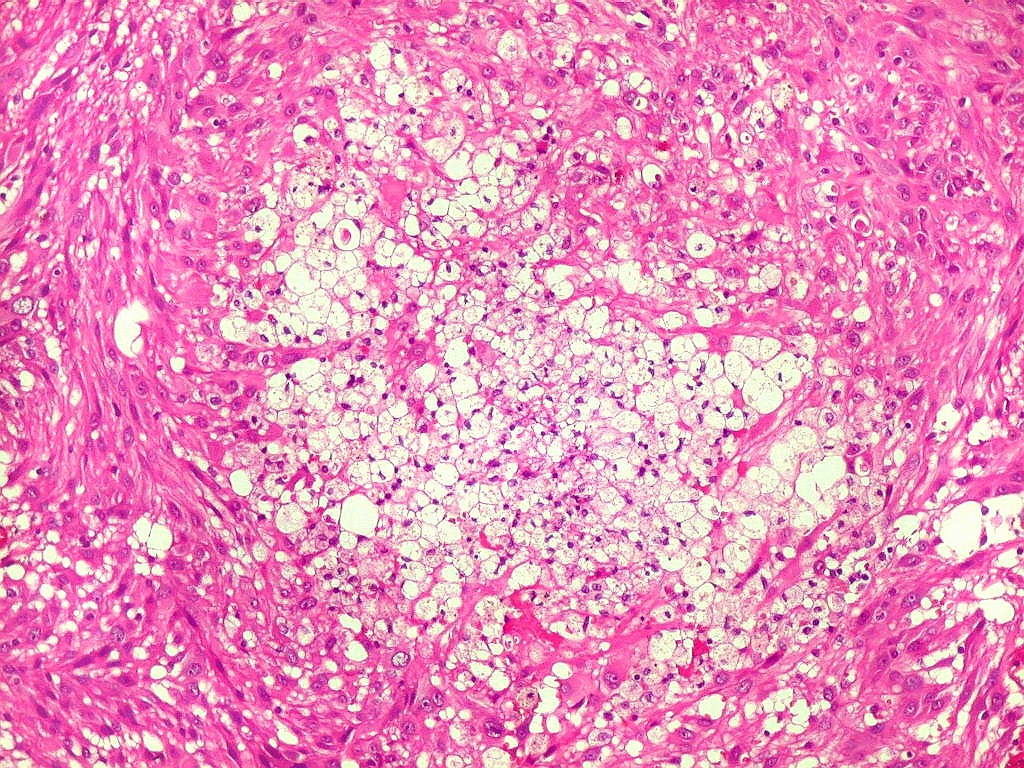
| Aspecto
geral do tumor.
A neoplasia apresentava aspectos variados em diferentes áreas. Predominavam células com aspecto astrocitário, com citoplasma abundante e núcleo excêntrico. Algumas ensaiavam arquitetura em feixes e distribuição perpendicular aos vasos. Estes eram finos, por vezes com espessamento fibroso da parede, mas sem proliferação endotelial. |
 |
 |
 |
 |
 |
 |
|
 |
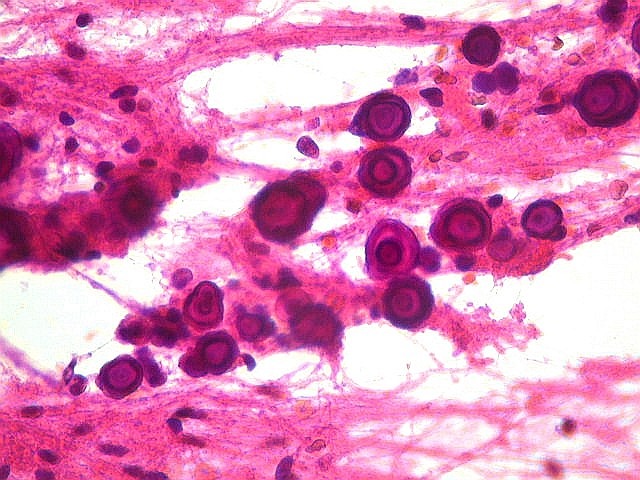
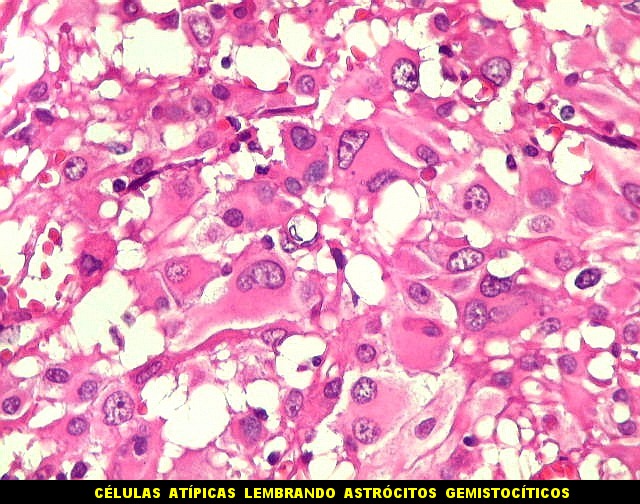
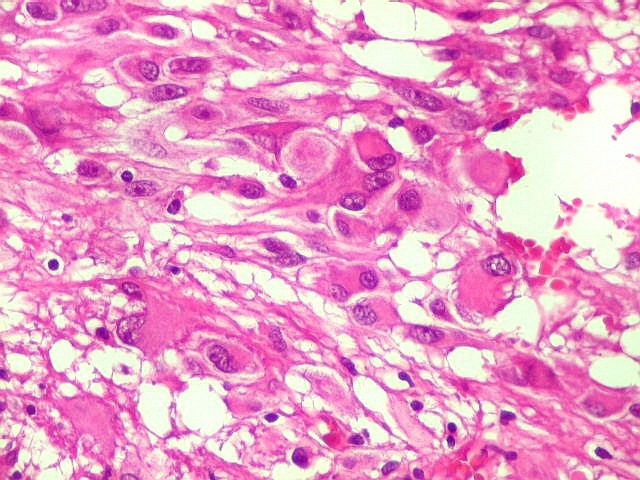
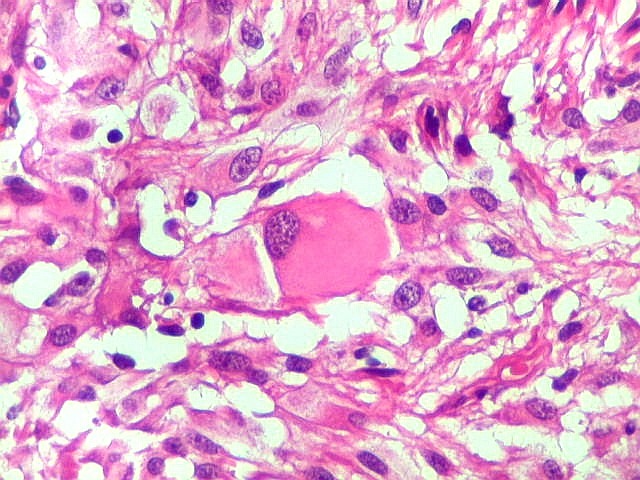
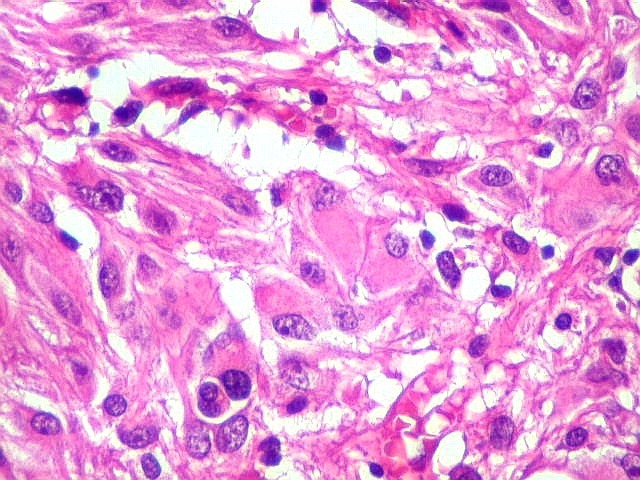
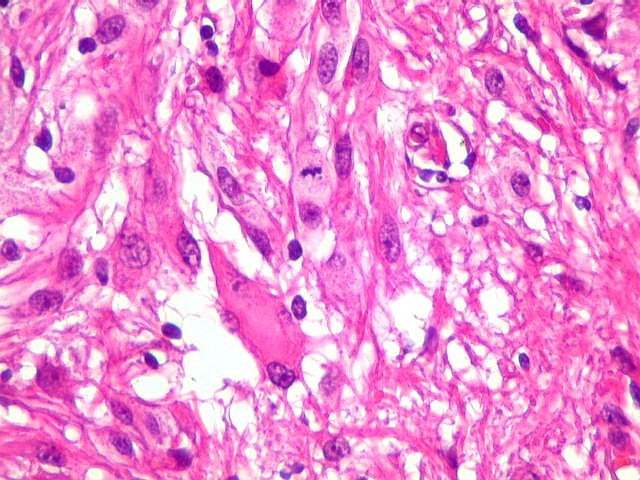
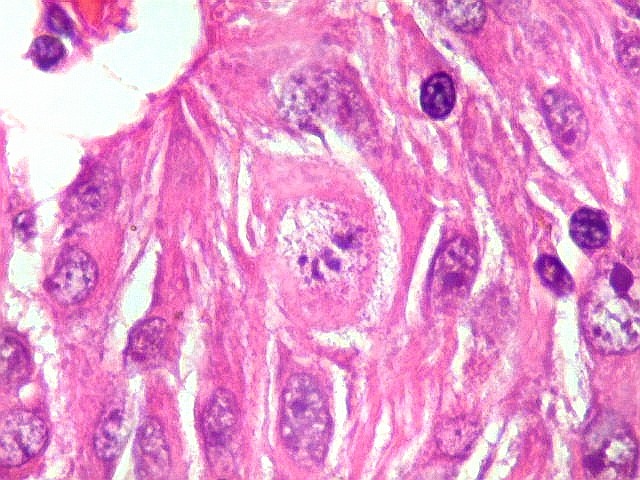
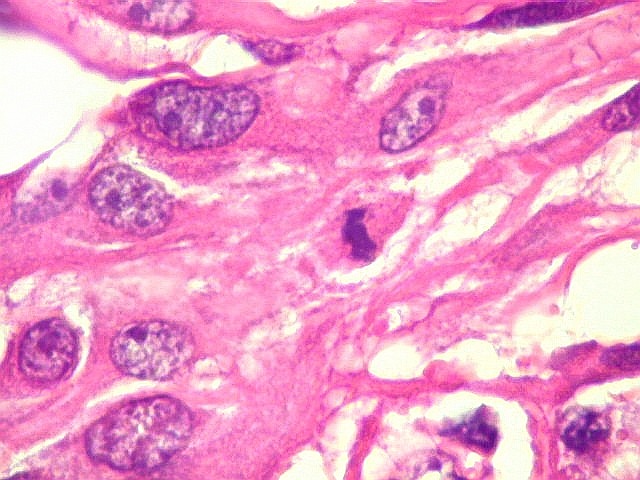
| Células atípicas lembrando astrócitos gemistocíticos. Em outras áreas predominavam células com as feições de astrócitos gemistocíticos - citoplasma abundante, róseo, homogêneo, e núcleo excêntrico. Havia freqüente bi- ou até trinucleação. Os núcleos eram volumosos, com cromatina bem distribuída e nucléolos proeminentes. | |
 |
 |
 |
 |
 |
 |
 |
|
 |
 |
 |
 |
 |
 |
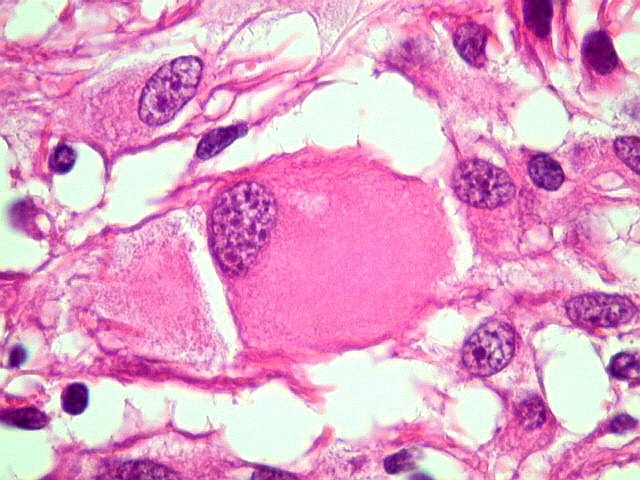
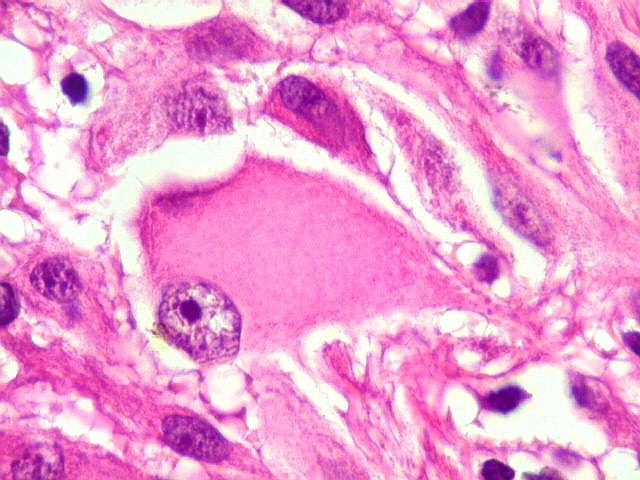
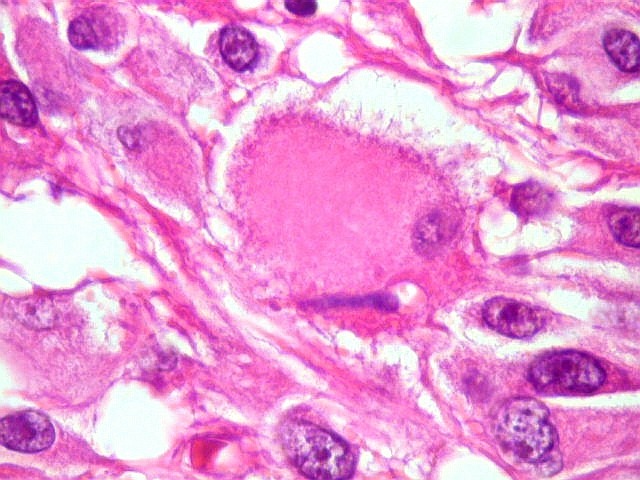
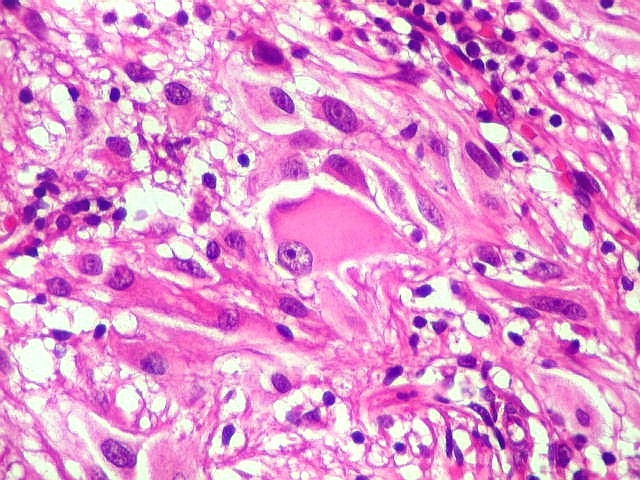
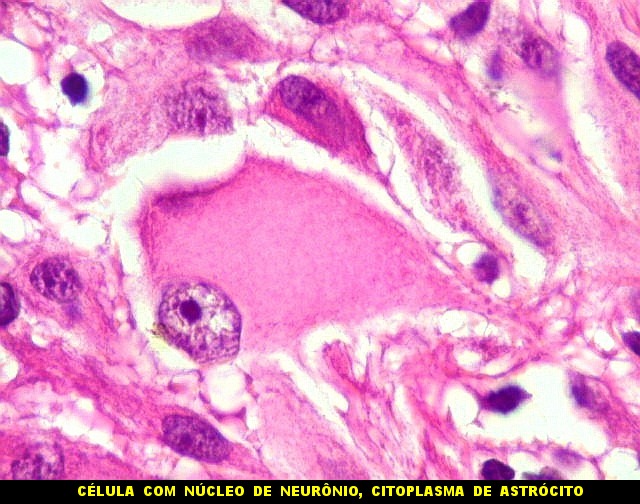
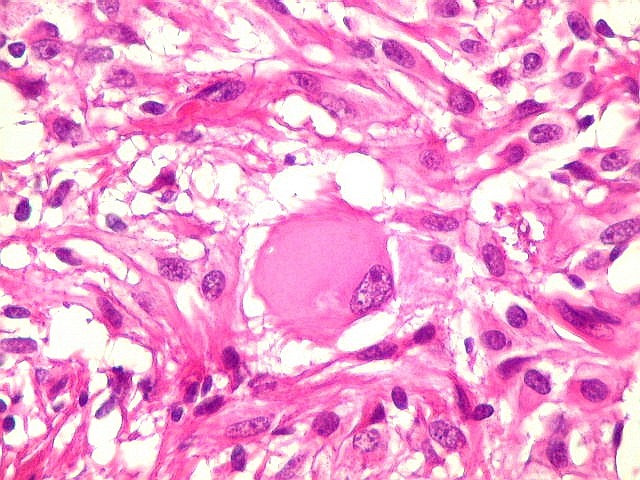
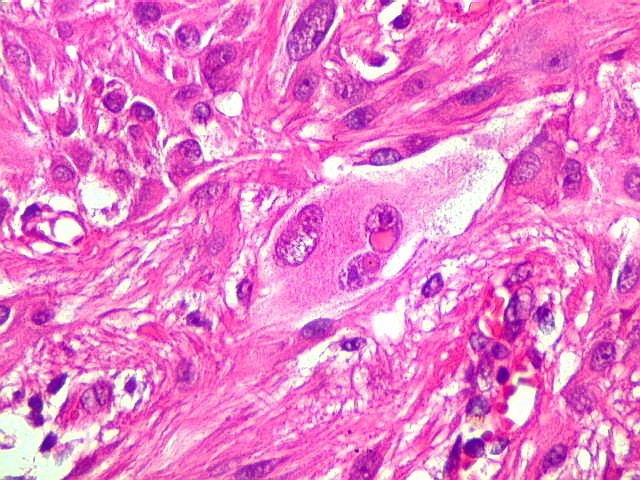
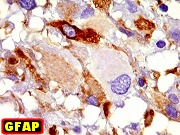
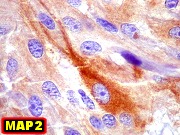
| Células com corpo de astrócito, núcleo lembrando neurônio. O caráter vesiculoso de alguns núcleos, com nucléolos muito evidentes, lembrava o aspecto de neurônios, embora o corpo celular fosse reminiscente de um astrócito gemistocítico. Várias células foram positivas para marcadores neuronais, como neurofilamento, cromogranina e MAP2 (clique para exemplos). Na literatura, esta dicotomia é reconhecida (texto), apoiando o caráter glioneuronal do astrocitoma subependimário de células gigantes. | |
 |
 |
 |
 |
 |
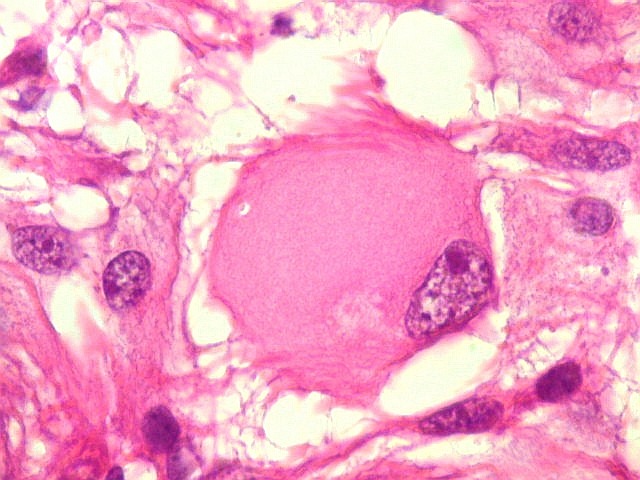 |
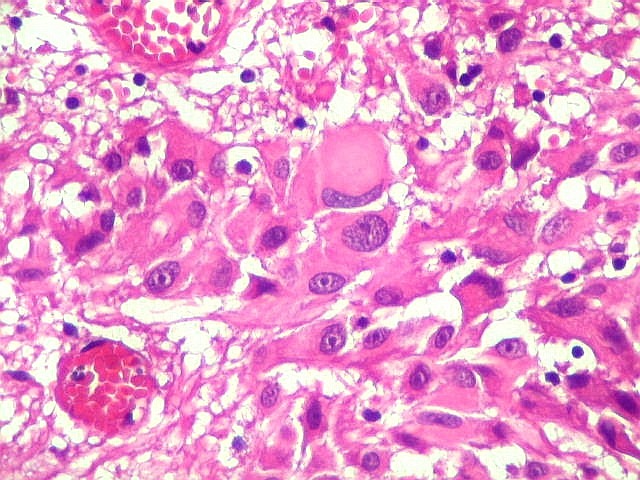
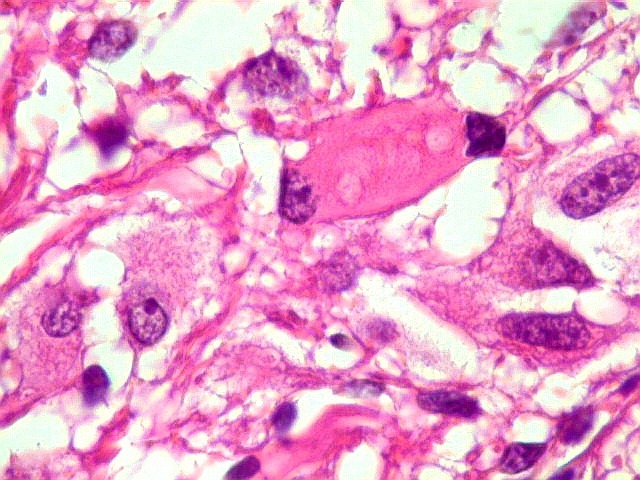
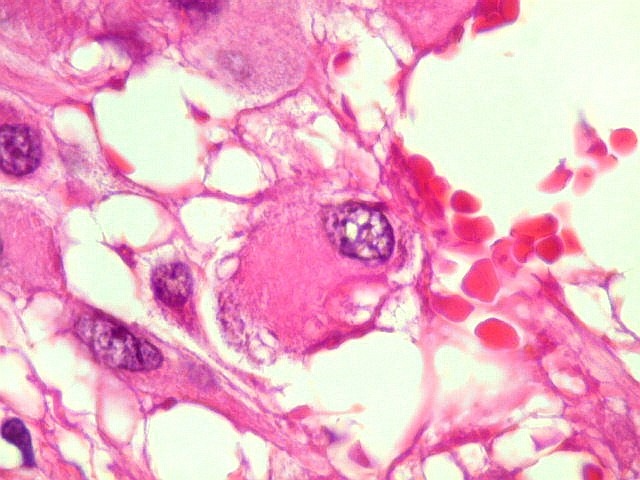
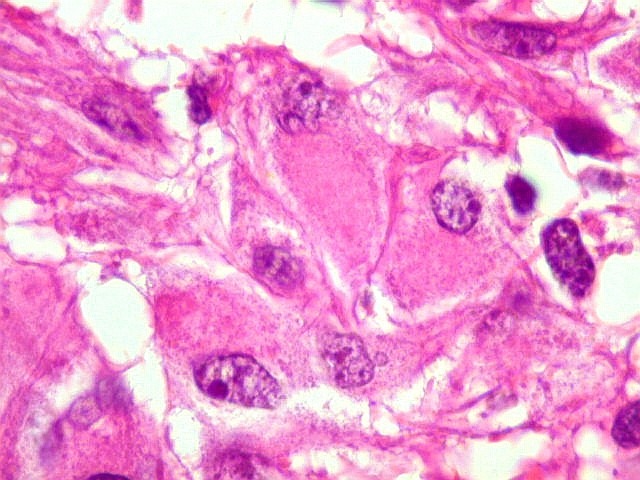
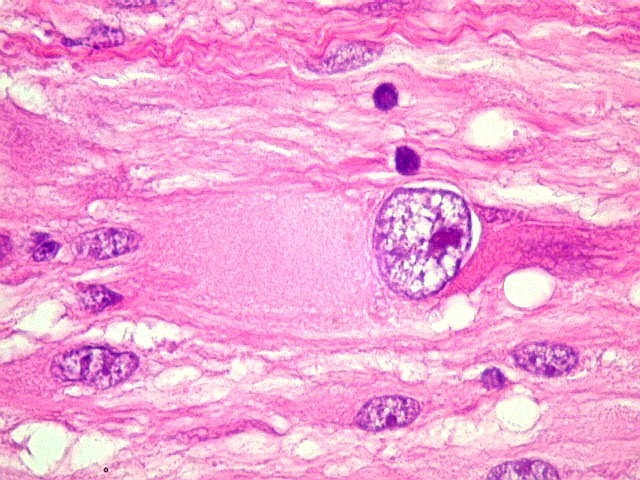
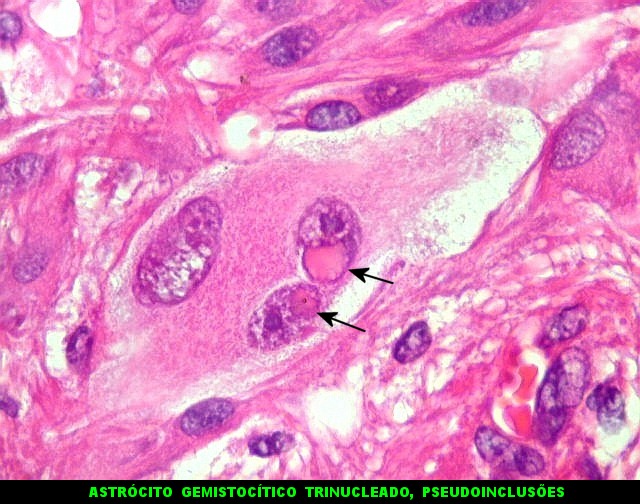
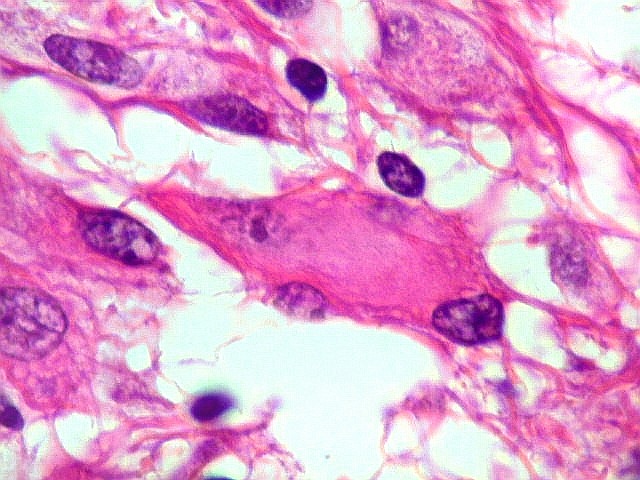
| Células multinucleadas. A célula multinucleada abaixo apresenta pseudoinclusões intranucleares, que aparecem como áreas homogêneas em róseo. Provavelmente se tratam de citoplasma insinuado em dobras da membrana nuclear. | |
 |
 |
 |
 |
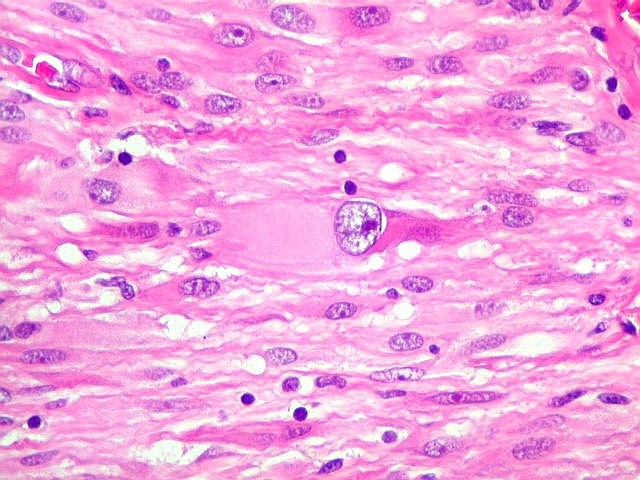
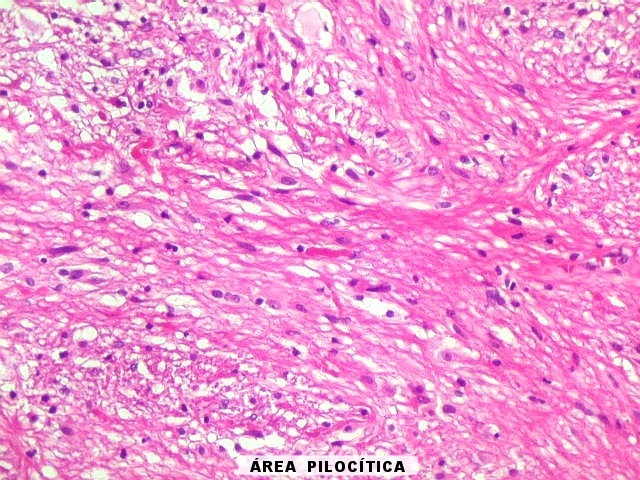
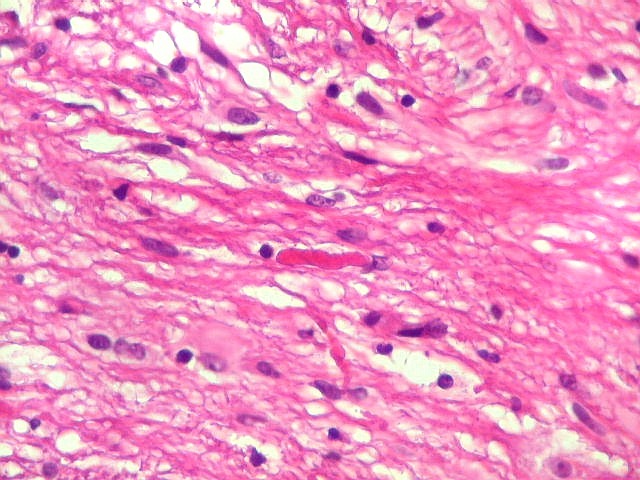
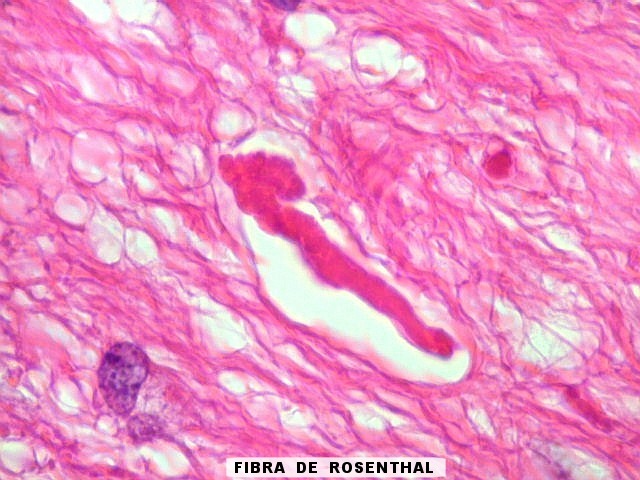
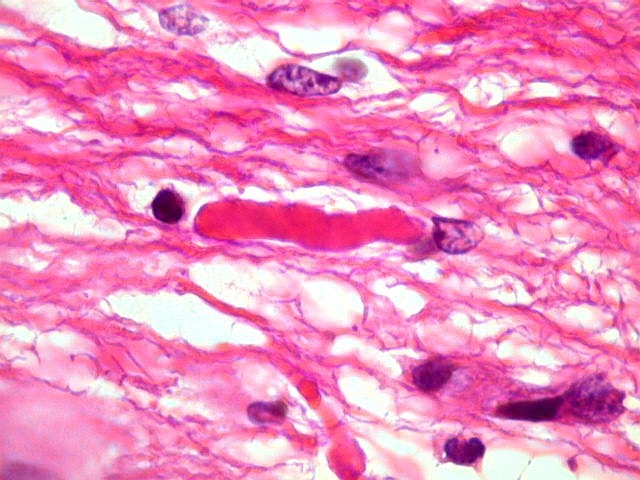
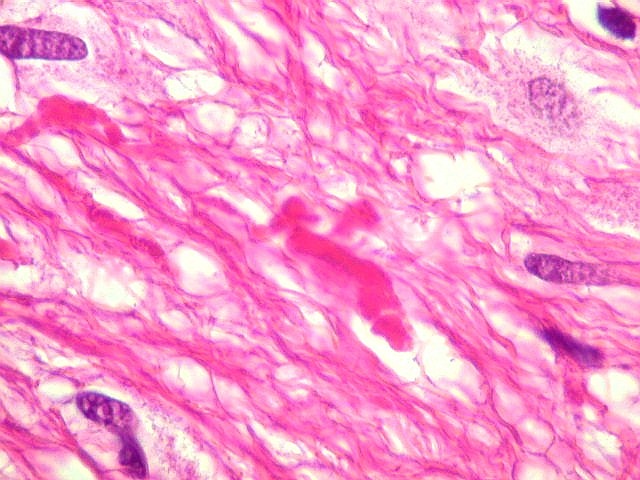
| Áreas pilocíticas com fibras de Rosenthal. Mais raramente, observavam-se áreas de células alongadas, com prolongamentos finos e de aspecto fibrilar, arranjadas em feixes. Algumas fibras de Rosenthal foram encontradas de permeio. O aspecto lembra o observado nos astrocitomas pilocíticos. | |
 |
 |
 |
 |
 |
 |
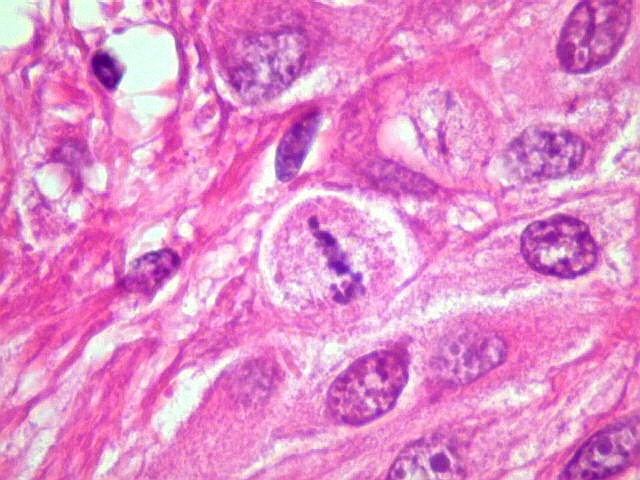
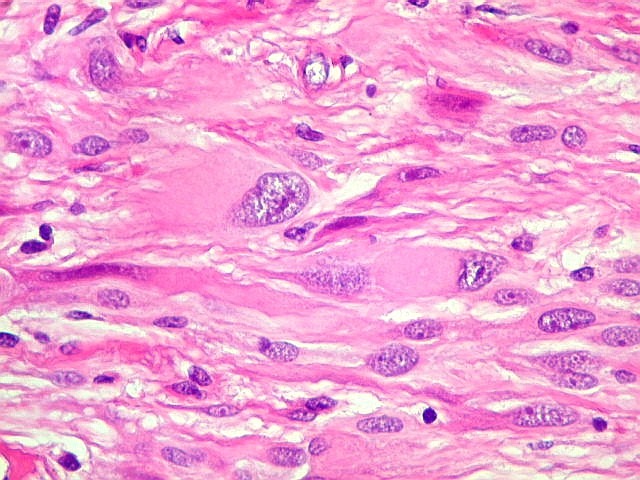
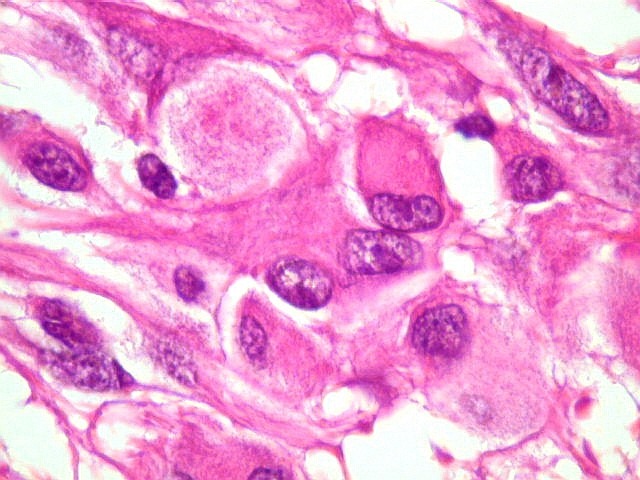
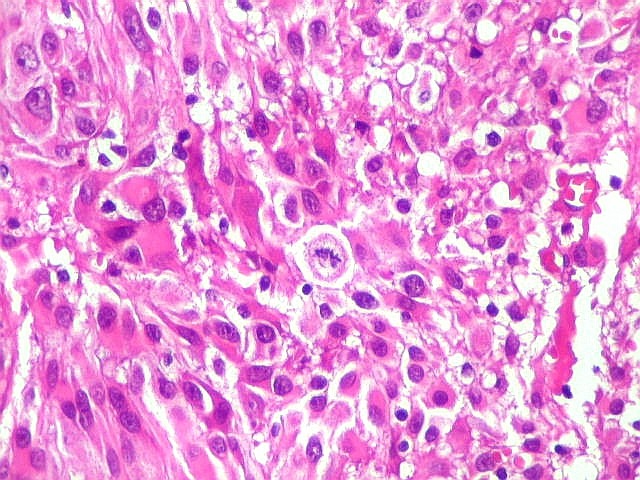
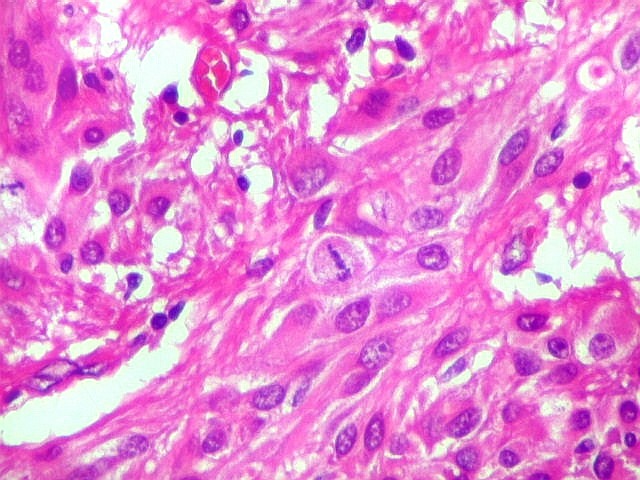
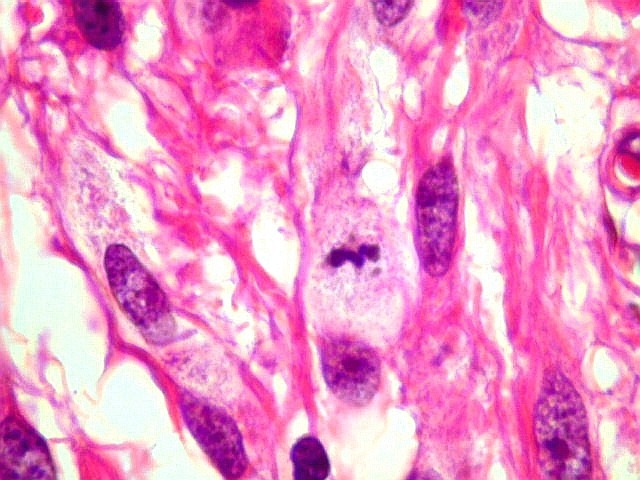
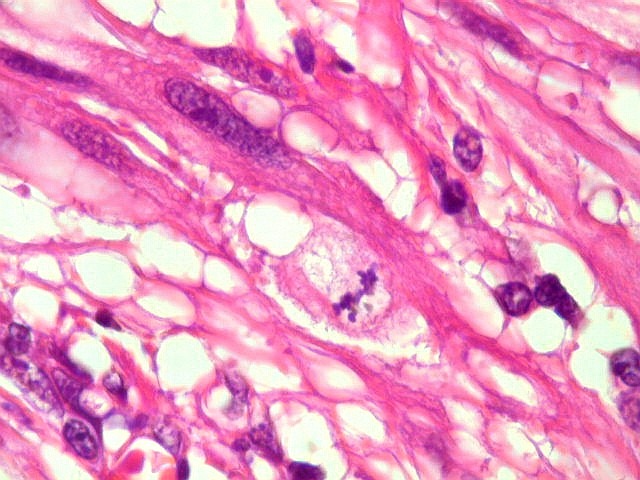
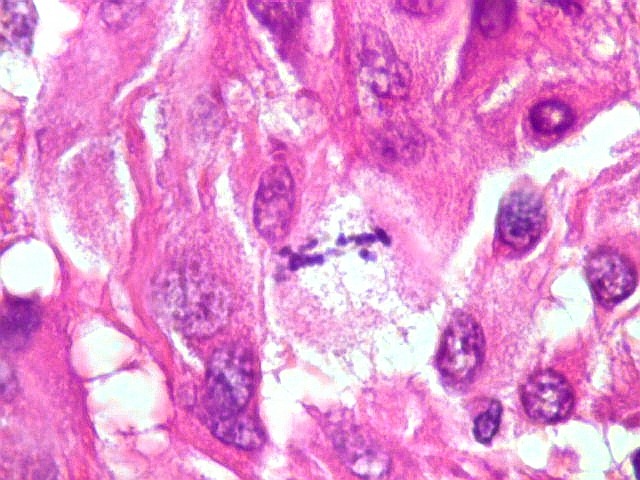
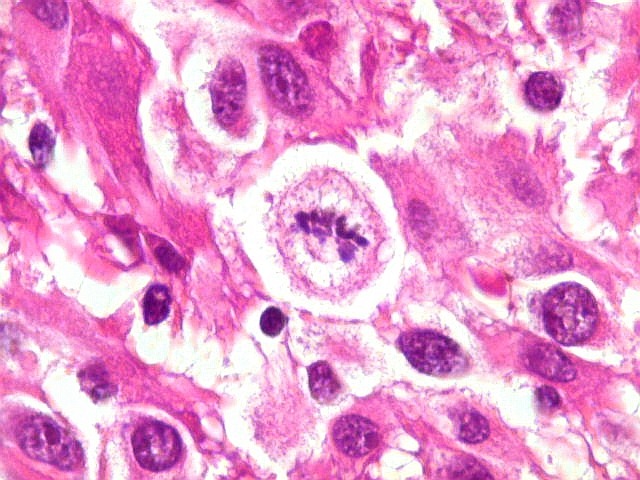
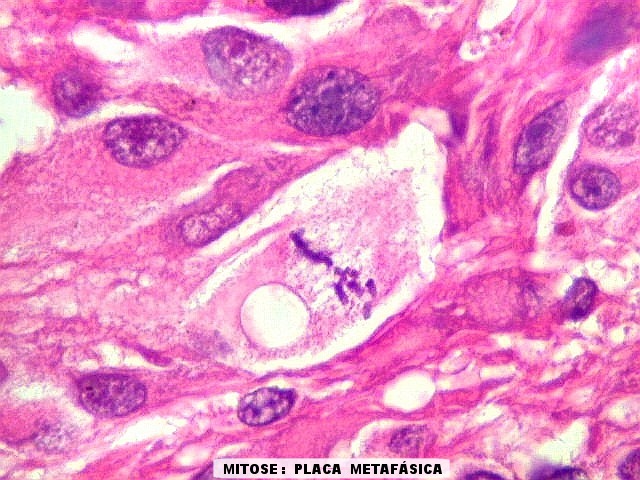
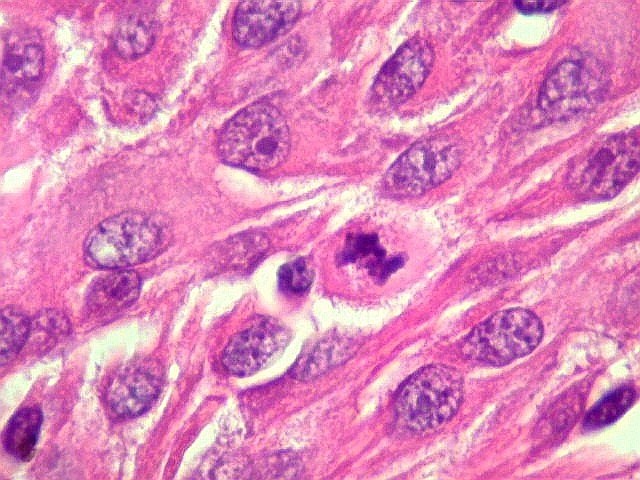
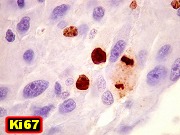
| Mitoses.
Figuras de mitose, todas típicas, foram notadas esparsamente pelo tumor. Seu número provavelmente ficava abaixo de 1% (não foram contadas). Pareciam predominar nas células globosas lembrando gemistócitos. O encontro de mitoses não é considerado sinal de malignidade ou de mau prognóstico nos astrocitomas subependimários de células gigantes, considerados grau I na classificação da OMS. |
 |
 |
|
 |
 |
 |
 |
 |
 |
 |
 |
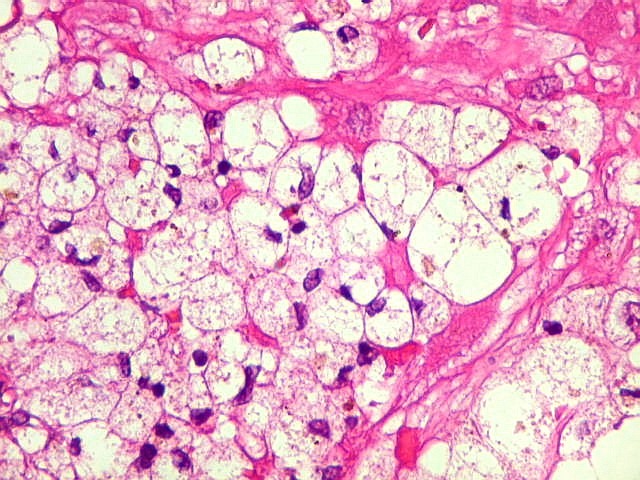
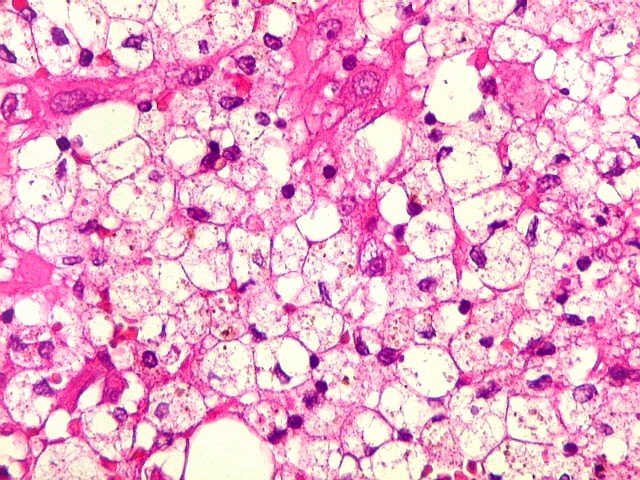
| Macrófagos
xantomatosos.
Em algumas pequenas áreas observavam-se acúmulos de macrófagos xantomatosos, provavelmente em processo de fagocitose de debris de células neoplásicas degeneradas. Não devem ser confundidos com as células xantomatosas do xantoastrocitoma pleomórfico, um tumor que pode levantar problemas de diagnóstico diferencial com o astrocitoma subependimário de células gigantes. As células xantomatosas do xantoastrocitoma são astrócitos, não macrófagos. Para mais sobre o xantoastrocitoma pleomórfico clique: imagem, patologia, texto. |
 |
 |
 |
|
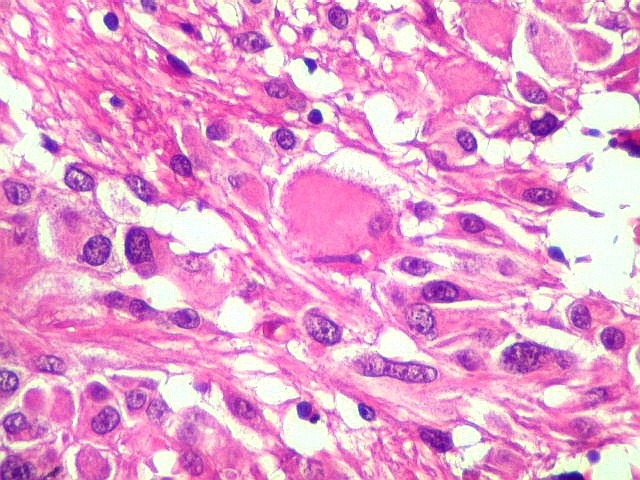
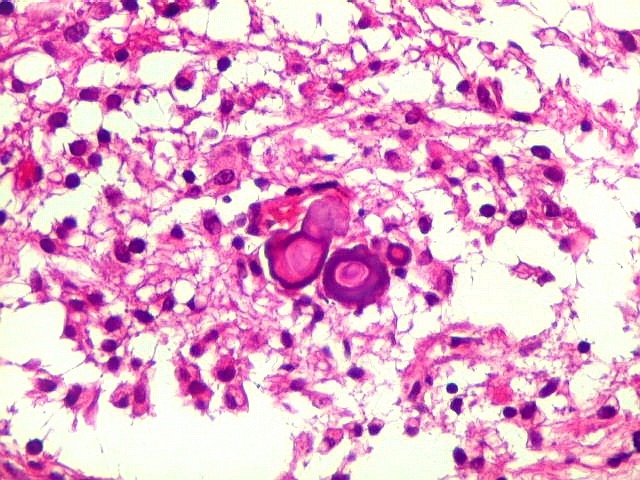
| Calcificações. Eram esparsas pelo tumor, na forma de concreções calcáreas, às vezes concêntricas. São uma feição característica do astrocitoma subependimário de células gigantes, freqüentemente detectáveis na tomografia computadorizada (clique para outros casos). No espécime usado para esfregaço eram muito mais abundantes. | |
|
 |
 |
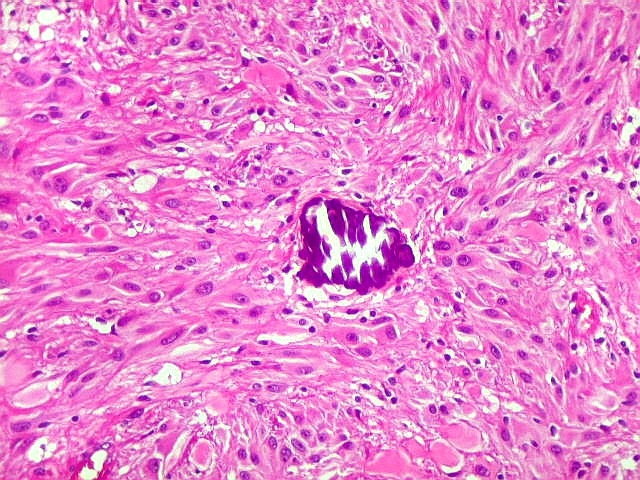 |
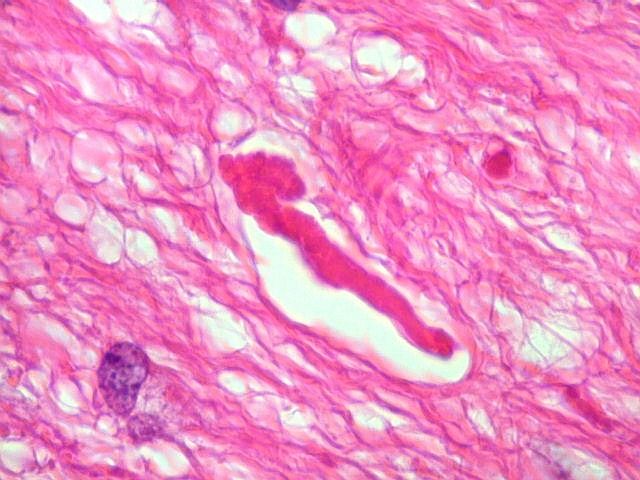
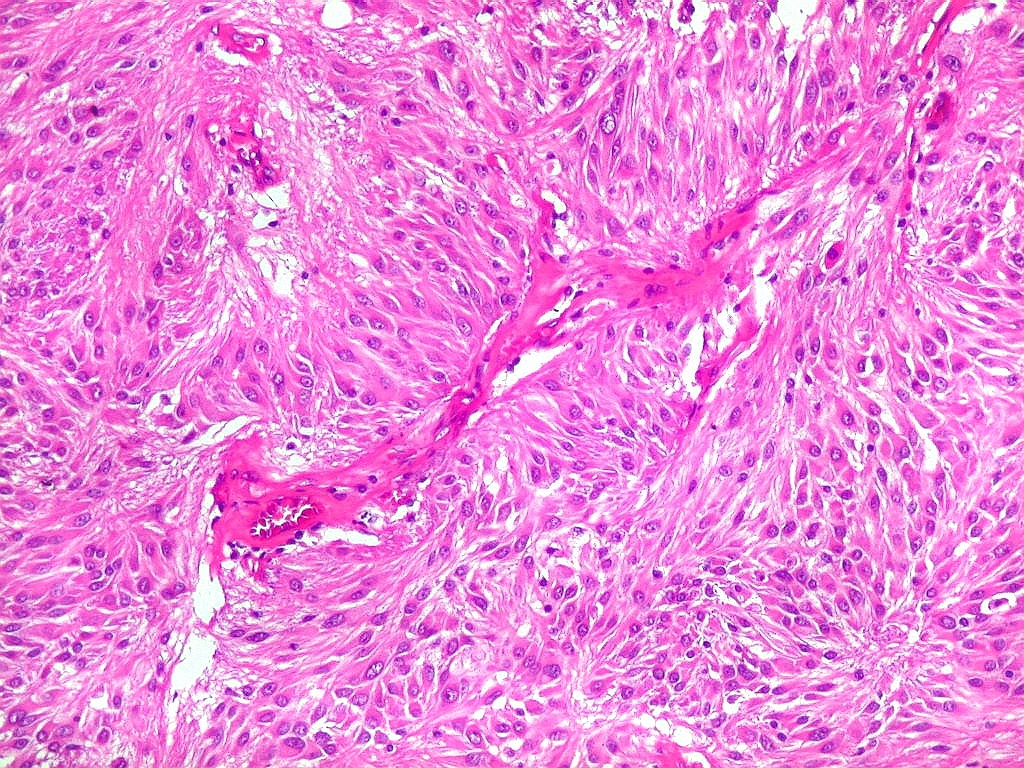
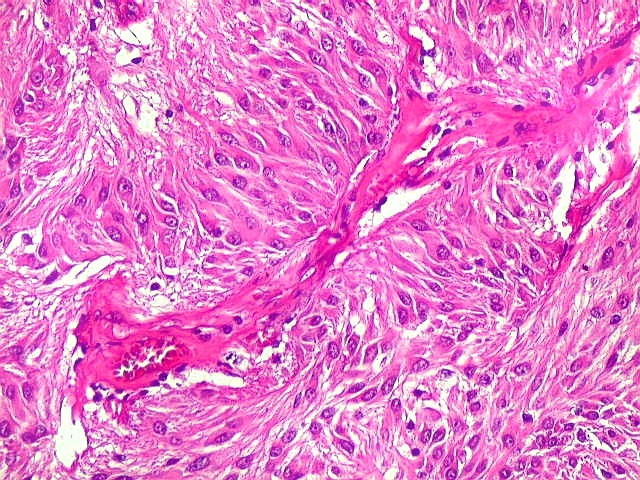
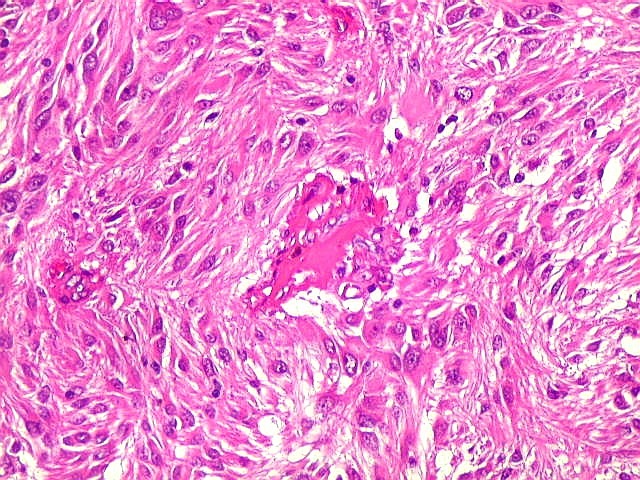
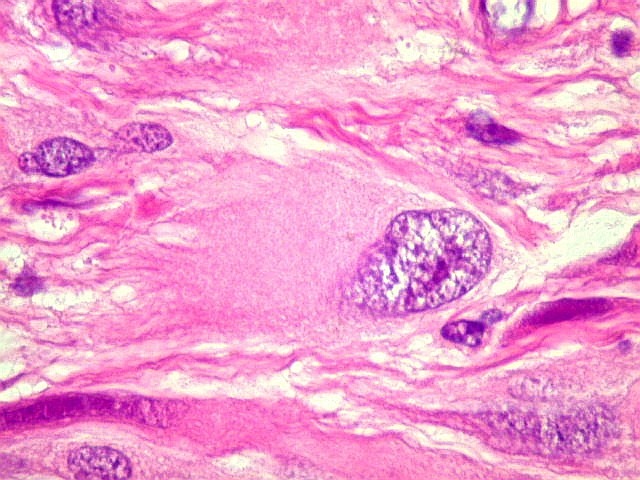
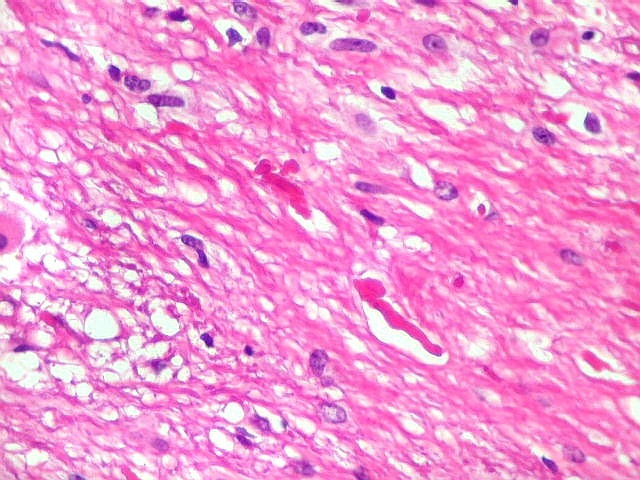
| Anormalidades
vasculares.
Aqui e acolá foram observados vasos aberrantes, com múltiplas luzes, paredes espessas e hialinizadas. Não havia, contudo, proliferação endotelial ou pseudoglomérulos, como encontrado nos gliomas malignos. Alguns vasos pareciam originar-se da reorganização de trombos. |
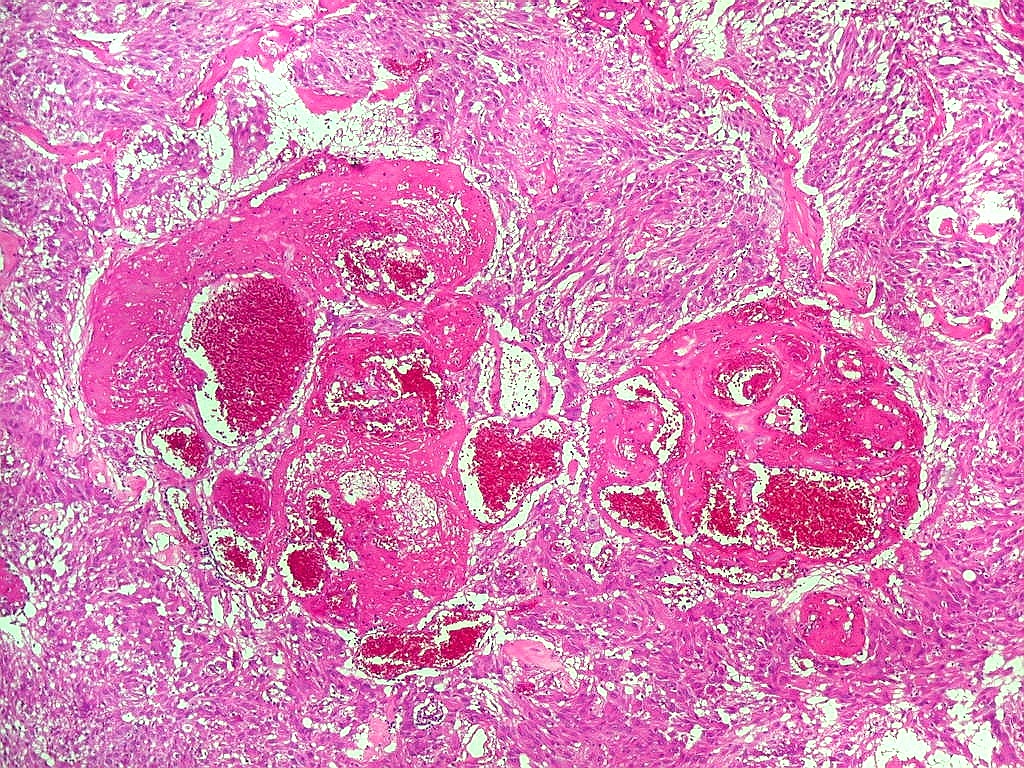 |
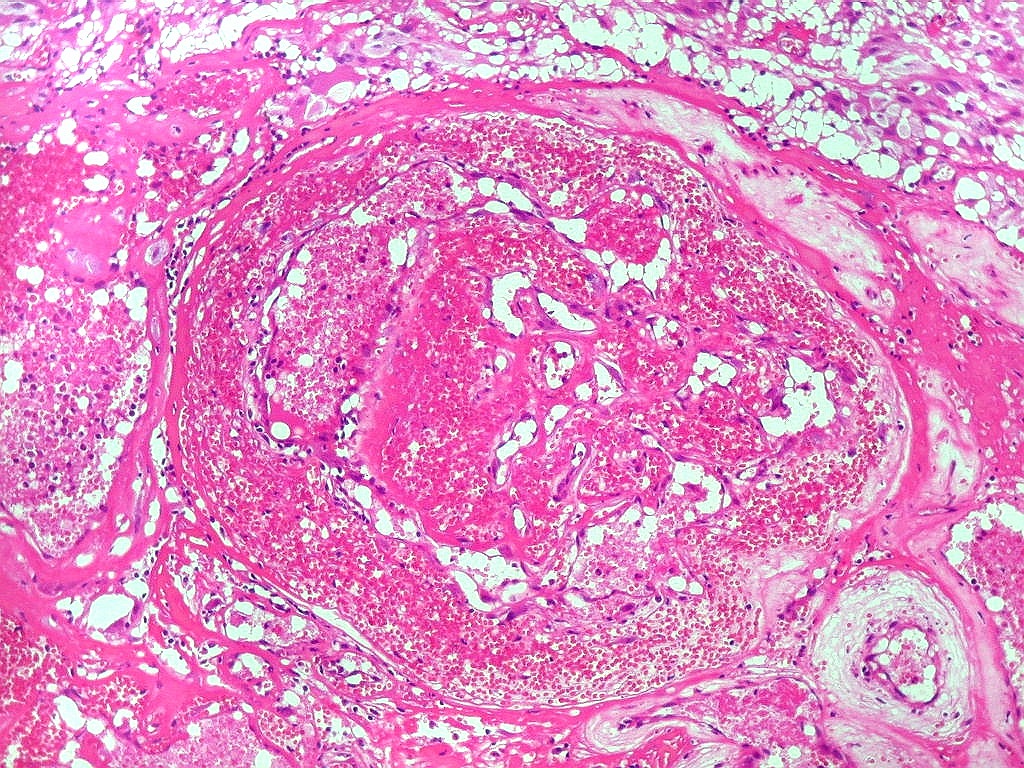 |
| Comentário.
Este
caso foi diagnosticado como astrocitoma subependimário de células
gigantes (ASECG) associando as feições de neuroimagem,
histopatologia
e imunohistoquímica. Na imagem,
o tumor localizava-se na parede do ventrículo lateral direito, com
calcificações grosseiras na sua porção inferior,
era bem delimitado e impregnava-se fortemente por contraste. A histologia
é compatível com ASECG, de um tumor astrocitário com
notável polimorfismo celular e nuclear, mas sem as feições
agressivas de um glioma difuso de alto grau. Poderia colocar-se o diagnóstico
diferencial com xantoastrocitoma pleomórfico (PXA), que também
apresenta pleomorfismo. Contudo, a topografia do PXA é superficial,
em relação com as leptomeninges, e freqüentemente cístico,
o que não concorda com os achados radiológicos. A imunohistoquímica
apóia ASECG e está de acordo com a literatura. Particulares
dignos de menção são a positividade para GFAP em apenas
parte das células, e a expressão de antígenos neuronais,
sugerindo uma natureza híbrida glioneuronal do tumor.
É clássico que o ASECG seja parte do quadro da esclerose tuberosa (ocorre em 6 a 18 % dos casos daquela) e discute-se sua existência fora desta entidade genética. O presente paciente não apresentava nenhum dos outros estigmas da síndrome, no exame físico, ressonância e tomografia de crânio, e exames de outros aparelhos. Pode ser que futuramente venha a apresentar, ou venham a ser descobertas outras feições da ET. Ou, que no presente caso, o ASECG seria a expressão única da síndrome (ficaria, segundo os critérios diagnósticos atuais, como 'possível ET'). Estudos genéticos podem ser realizados no sentido de detectar mutações nos genes TSC1 e TSC2. De qualquer modo, do ponto de vista morfológico e radiológico, cremos que o diagnóstico de ASECG se impõe e não temos alternativas como diagnóstico diferencial. Na literatura, há raros relatos de casos de ASECG fora da esclerose tuberosa. Para referências adicionais, ver:
|
| Astrocitoma
subependimário de células gigantes
Definição. Neoplasia astrocitária benigna, bem delimitada e de crescimento lento, originado nas paredes dos ventrículos laterais e composto por células grandes, atípicas, semelhantes a astrócitos. Quase sempre ocorrem nas proximidades do forâmen de Monro em crianças e adultos jovens. São raros (< 1% dos tumores intracranianos). É a neoplasia cerebral mais comum na esclerose tuberosa e discute-se sua ocorrência fora da doença. Incidência varia entre 6 e 14% dos pacientes com esclerose tuberosa confirmada e o tumor é um dos critérios maiores para o diagnóstico da síndrome. É importante reconhecer estes tumores para evitar confusão com neoplasias mais agressivas (como os astrocitomas difusos de alto grau). Graduação. Correspondem a grau I da OMS. Idade. Tipicamente nas duas primeiras décadas (variando de 2 a 30 anos, média 13 anos). Há raras descrições em recém-nascidos ou casos congênitos. Clínica. A maioria dos pacientes apresenta piora do quadro epiléptico ou sintomas de hipertensão intracraniana. Calcificações intracranianas podem ser observadas em radiografias simples ou TC. Imagem. Lesões solitárias e circunscritas nos ventrículos laterais, geralmente na região do forâmen de Monro, podendo envolver o III ventrículo adjacente. Tamanho entre 1 a 6 cm. Não mostram feições invasivas, mesmo quando volumosos. Em TC têm freqüentemente calcificações grosseiras. Em RM têm iso ou hipossinal em T1, iso ou hipersinal em T2 e impregnam-se fortemente por contraste. Obstrução liquórica pode levar a hidrocefalia e transudação transpendimária. Micro. Lesões são circunscritas, freqüentemente calcificadas. São compostas por células grandes, globosas, citoplasma róseo e vítreo, parecendo astrócitos gemistocíticos. Há grande variação morfológica, com células menores, alongadas em estroma fibrilar, ou células epitelióides. As células se arranjam em fascículos multidirecionados, ou em lóbulos separados por septos conjuntivos. Núcleos mostram cromatina finamente granulosa, com nucléolos proeminentes. Pode haver considerável polimorfismo nuclear, células gigantes multinucleadas e atividade mitótica, sem implicar em mau prognóstico. Ocasional proliferação endotelial ou necrose também não têm significado adverso. Há boa delimitação com o cérebro adjacente, sem infiltração. Índice Ki-67 é baixo, entre 1,5 e 7,4 %. Pode haver extensa calcificação. Imunohistoquímica. Revela fenótipo misto glioneuronal. As células mostram imunorreatividade variável para GFAP e S-100. A positividade para GFAP pode ser surpreendentemente escassa ou focal. S-100 é mais uniformemente positivo. Há positividade para beta-tubulina classe III associada a neurônios, que parece ser o epítopo neuronal mais expressado. Proteína de neurofilamento (NF) é restrita a alguns prolongamentos celulares e poucas células ganglionares (= células com diferenciação neuronal). Reatividade para SNF é rara. Pode haver expressão de cromogranina. Diagnóstico diferencial radiológico. Tumores que ocorrem no sistema ventricular próximos ao forâmen de Monro incluem o neurocitoma central, subependimoma, meningioma, tumores do plexo coróide e tumores germinativos. Todos são facilmente distinguíveis do astrocitoma subependimário de células gigantes pela histologia. Diagnóstico diferencial histológico. Pode haver confusão com :
Definição. Grupo de doenças autossômicas dominantes, caracterizadas por hamartomas e lesões neoplásicas benignas afetando o sistema nervoso central (SNC) e vários tecidos não neurais. As principais manifestações cerebrais incluem os hamartomas corticais ou túberes, hamartomas glio-neuronais subcorticais, nódulos gliais subependimários e o astrocitoma subependimário de células gigantes. As principais manifestações extraneurais incluem angiofibromas cutâneos (adenoma sebáceo de Pringle), peau chagrin, fibromas subungueais, rabdomiomas cardíacos, pólipos intestinais, cistos viscerais, linfangioleiomiomatose pulmonar e angiomiolipomas renais. Peau de chagrin, shagreen patch, cútis áspera. Trata-se de áreas em placa de pele espessada e finamente rugosa, que pode ser comparada à de uma laranja (orange peel), ou também a pele de porco (pigskin) ou de elefante (elephant hide), geralmente na região lombo-sacral. Podem ter de 1 a 10 cms. e correspondem a uma placa de fibrose subepidérmica. O termo deriva do original turco çagri (garupa de cavalo) e pode ser traduzido em português como chagrém. É um tipo de couro granulado, preparado sem curtir, usado em encadernações. Também: pele áspera de alguns tubarões e raias, usada como lixa ou abrasivo. Para imagens na internet, clique (1) (2) (3). Causa genética básica. Mutação no gene TSC1 no cromossomo 9q, ou no gene TSC2 no cromossomo 16p. Sinônimos. Doença de Bourneville, ou de Bourneville Pringle. Incidência. A incidência na população norte-americana seria de cerca de 1 : 5000. Admite-se afecção de 25.000 a 40.000 indivíduos nos Estados Unidos e 1 a 2 milhões no mundo. Prevalência 1/6000 nascimentos. Critérios para diagnóstico. O diagnóstico pode ser difícil devido à ampla variabilidade das alterações. A maioria dos pacientes já tem manifestações antes dos 10 anos. Sintomas neurológicos são os mais comuns e importantes, e incluem epilepsia, espasmos infantis, retardo mental e alterações do comportamento, como autismo. Os critérios abaixo foram definidos em consenso internacional em 1998 (incidências aproximadas entre parênteses). Critérios maiores. Angiofibromas
faciais (80 90%)
Critérios menores. Depressões
(pits) no esmalte dentário, múltiplas e distribuídas
ao acaso (30%)
O diagnóstico de esclerose tuberosa é baseado nos critérios acima e dado como definitivo, provável ou possível. Definitivo
: dois critérios maiores; ou um critério maior + 2
menores.
Túberes corticais, heterotopias na substância branca, e nódulos hamartomatosos subependimários, comparados classicamente a gotas de cera de vela, ocorrem em 90 a 100% dos casos de esclerose tuberosa. Em comparação, o astrocitoma subependimário de células gigantes é observado em 6 a 16% dos casos. Túberes corticais são detectáveis por TC ou RM e estão especialmente associados a epilepsia. Microscopicamente, consistem de células gigantes, semelhantes às do astrocitoma subependimário. Há também neurônios dismórficos, distúrbios da laminação cortical, gliose, calcificação de pequenos vasos e perda da mielina. As lesões são focais, com o córtex vizinho apresentando características normais. Os neurônios dismórficos e as células gigantes podem ser vistos em todas as camadas corticais e na substância branca adjacente. Os neurônios displásicos mostram orientação radial alterada, arborização dendrítica aberrante, acúmulo de fibrilas no pericário, e morfologia somática aberrante com citoplasma eosinófilo abundante (células em balão ou balloon cells). Os neurônios expressam suas proteínas próprias, mas têm feições citoarquiteturais de neurônios imaturos ou pouco diferenciados como diminuição das projeções axonais e da densidade das espinhas dendríticas. As células gigantes nos túberes corticais mostram uma heterogeneidade celular e molecular semelhante às observadas no astrocitoma subependimário de células gigantes. A coexistência de marcadores neuronais e gliais sugere origem mista glioneuronal. Muitas células gigantes em túberes expressam nestina (tanto mRNA como a proteína). Enquanto algumas expressam GFAP, outras com morfologia idêntica demonstram marcadores neuronais, como conexinas 26 e 32, neurofilamentos, classe III beta tubulina, MAP2 e alfa-internexina. Hamartomas corticais morfologicamente indistinguíveis de túberes podem ocorrer em epilepsias crônicas focais sem evidência clínica de esclerose tuberosa. Genética. Cerca de 50% dos pacientes com esclerose tuberosa têm história familial, o que indica alta taxa de novas mutações. Nas famílias afetadas a doença segue herança autossômica dominante com alta penetrância, mas considerável variação fenotípica. Estudos neurorradiológicos sugerem que parentes em primeiro grau podem ter sinais clínicos menores ou formas frustas da doença. Há dois loci associados à esclerose tuberosa, um no cromossomo 9q (TSC1) e outro no cromossomo 16p (TSC2), provavelmente genes supressores tumorais. Não há distinção fenotípica clara associada a cada um deles em separado, sugerindo que os dois atuariam na mesma via reguladora (os produtos de ambos interagiriam). Mutações em TSC2 são muito mais comuns que em TSC1. TSC1 codifica a proteína hamartina de 130 kD, fortemente expressada no cérebro, coração e rins (todos órgãos afetados na esclerose tuberosa). Sua expressão topográfica sobrepõe-se à da tuberina (180 kD), produto do gene TSC2. Por enquanto não se demonstraram correlações entre genótipo e fenótipo nas mutações destas proteínas. Pacientes com mutações de TSC1 tendem a ter doença um pouco mais leve que os com TSC2, quando comparados em idades semelhantes. Mutações de TSC1 responderiam por 10 a 15% dos casos, de TSC2 por 55 a 65%. As duas proteínas interagem no citoplasma formando um complexo que funciona como supressor tumoral. A penetrância das mutações de ambos genes é de 100%, mas o fenótipo é altamente variável. Genótipo não permite predizer o fenótipo, mesmo um uma só família, indicando atuação de outros fatores. Gêmeos monozigóticos podem ter manifestações bem diferentes. Embora lesões focais associadas a epilepsia crônica mostrem alterações celulares praticamente indistinguíveis das dos túberes da esclerose tuberosa, não foram detectadas mutações dos genes TSC1 e TSC2 nestas lesões malformativas esporádicas. Os achados não apóiam que estes pacientes sejam portadores de uma forma frusta da esclerose tuberosa. Fontes.
|
| Para mais imagens deste caso, comentário, textos ASECG, ET. | |||
| TC, RM | IH marcadores gliais | IH marcadores neuronais | IH Nestina, EMA, CD34, Ki-67 |
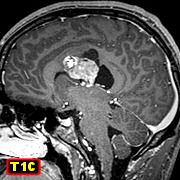 |
 |
 |
 |
| Textos: ASECG, esclerose tuberosa (1) (2) | Banco de Imagens: Neuropatologia da esclerose tuberosa | Características de imagem da esclerose tuberosa | ASECG, mais casos: Imagem, Patologia |
| Neuropatologia
- Graduação |
Neuropatologia -
Estudos de casos |
Neuroimagem
- Graduação |
Neuroimagem -
Estudos de Casos |
Roteiro
de aulas |
Textos
de apoio |
Correlação
Neuropatologia - Neuroimagem |
| Índice alfabético - Neuro | Adições recentes | Banco de imagens - Neuro | Textos ilustrados | Neuromuscular | Patologia - outros aparelhos | Pages in English |
|
|
|
|
{kind=link}