|
|
(angiomatose encéfalo-trigeminal) Texto e ilustrações com links para casos |
|
|
|
(angiomatose encéfalo-trigeminal) Texto e ilustrações com links para casos |
|
|
Síndrome de Sturge-Weber. Sinônimos angiomatose encéfalo-trigeminal ou meningofacial, síndrome de Sturge-Weber-Dimitri. Definição. Doença do desenvolvimento caracterizada por angiomatose da face, da coróide ocular e leptomeninges, sem fundo genético reconhecido. Ambos sexos são igualmente acometidos. A grande maioria dos casos é esporádica e não se descobriu um mecanismo de herança, embora sejam registrados casos familiais. Clínica. A lesão de face pode ser parcial ou total (chamada nevus flammeus, ou angioma em vinho do porto [port-wine nevus]). Pode haver acometimento de um, ou de ambos lados da face, envolvendo mais a porção média. Corresponde ao território do N. trigêmeo, mas a associação é fortuita não quer dizer que este nervo participe da patogênese da doença. Presente já ao nascimento, a lesão facial é constituída por abundantes vasos semelhantes a capilares, e não se altera com o avançar da idade. Descrevem-se casos em que a lesão facial está ausente na vigência das manifestações intracranianas típicas (estas, presentes em 100% dos casos, já que são critério de definição da doença). A lesão cerebral pode manifestar-se por convulsões, hemiparesia, hemianopsia, ou retardo mental. Os pacientes desenvolvem-se normalmente até o início de crises focais ou generalizadas. Espasmos da infância aparecem em cerca de 90% dos casos durante o primeiro ano de vida, seguidos por crises tônicas, atônicas ou mioclônicas, que vão se tornando progressivamente refratárias à medicação. São acompanhadas por hemiparesia progressiva em cerca de 30% dos casos, freqüentemente associada a hemianopsia homônima. A maioria dos pacientes tem retardo mental. Anatomia patológica. A principal alteração cerebral são abundantes pequenos vasos dilatados (capilares e vênulas), geralmente restritos à pia-máter de um dos hemisférios cerebrais. As porções posteriores dos hemisférios cerebrais são preferencialmente acometidas, com predomínio nos lobos occipitais, seguindo-se os temporais e parietais, os frontais relativamente poupados. Estruturas da fossa posterior (cerebelo, tronco cerebral) não participam, presumivelmente porque sua origem embriológica é diferente. As artérias são menos envolvidas e tendem a sofrer fibrose. Patogênese.
É pouco conhecida.
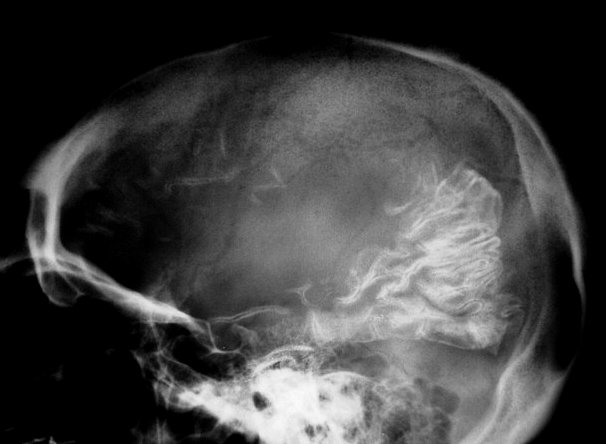
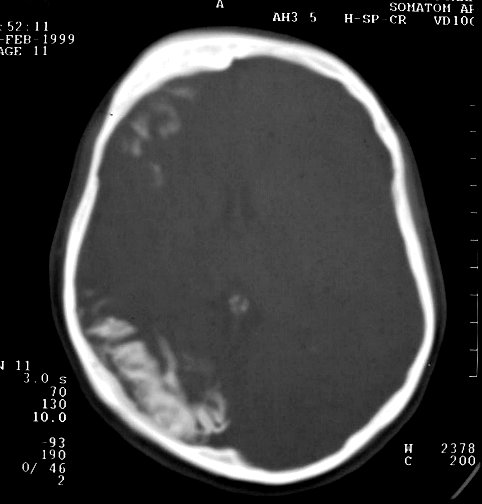
Calcificações
corticais são outra importante manifestação
patológica da síndrome e ocorrem só nas áreas
cerebrais subjacentes ao angioma (regiões posteriores de um dos
hemisférios). Começam na substância branca subcortical
e depois aparecem no córtex, predominantemente nas camadas II e
III. Podem ser bilaterais em 20% dos casos. Provavelmente, devem-se à
isquemia crônica por deficiência da drenagem venosa. As calcificações
são o achado mais comum da síndrome de Sturge-Weber em tomografias
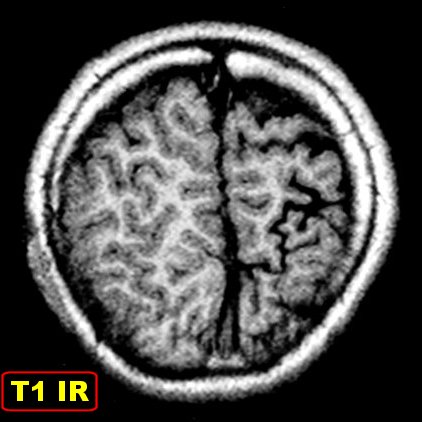
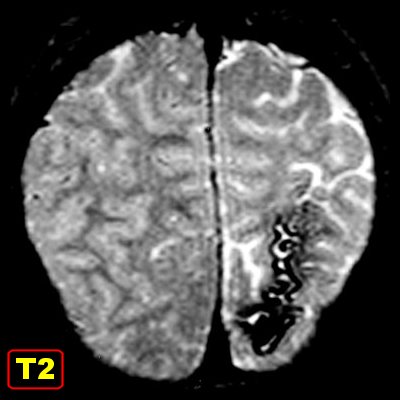
(TC). Na ressonância magnética (RM), podem ser difíceis
de visualizar em T1; são melhor notadas em T2 como uma fita de hiposinal
no córtex.
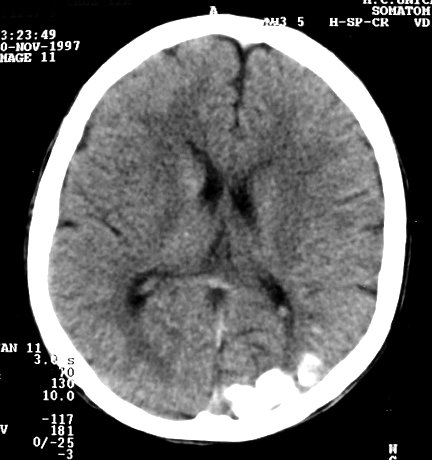
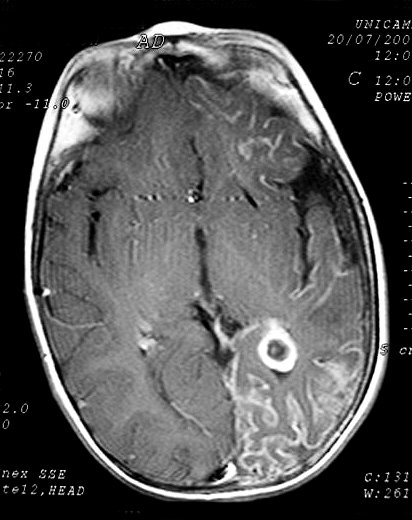
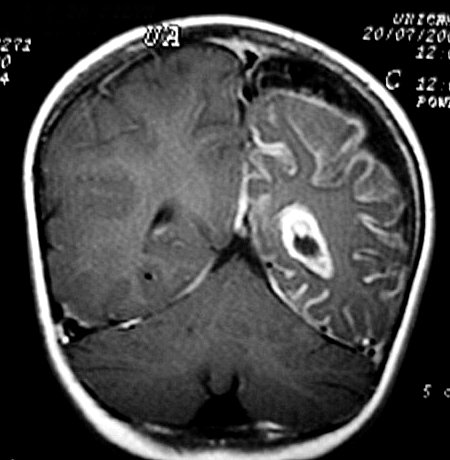
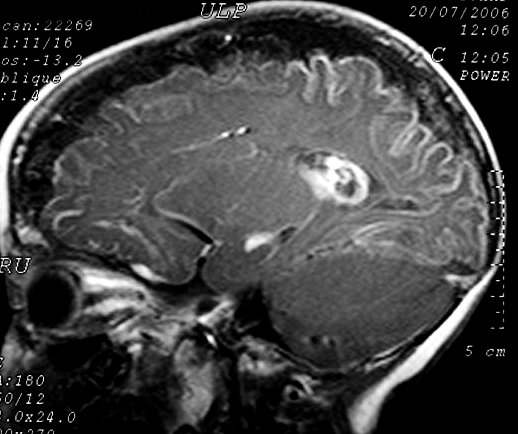
Neuroimagem. RM em T1 com contraste é o método mais acurado para mostrar a extensão do angioma pial, pois há intenso realce pelo gadolínio dos capilares meníngeos dilatados. Na infância, antes do aparecimento das calcificações, a captação de contraste pode ser observada também na TC. Com o tempo, as calcificações mascaram a captação. Com contraste, as áreas captantes na RM cobrem a superfície dos giros e preenchem os sulcos. O grau e extensão do angioma estão relacionados ao prognóstico, assim RM é essencial para planejar cirurgia em pacientes com convulsões de difícil controle. Em casos muito avançados, com cérebro muito atrófico e calcificado, pode haver pouca impregnação, possivelmente por trombose dos vasos angiomatosos. É
comum aumento de volume do plexo coróide, que se correlaciona com
a extensão do angioma leptomeníngeo. Há forte captação
de contraste pelo plexo ipsilateral ao angioma. Nas seqüências
com TR longo (T2, FLAIR), o plexo afetado está hiperintenso em relação
ao parênquima cerebral. Há uma hiperplasia do plexo,
possivelmente relacionada ao aumento do fluxo venoso pelo hemisfério
afetado.
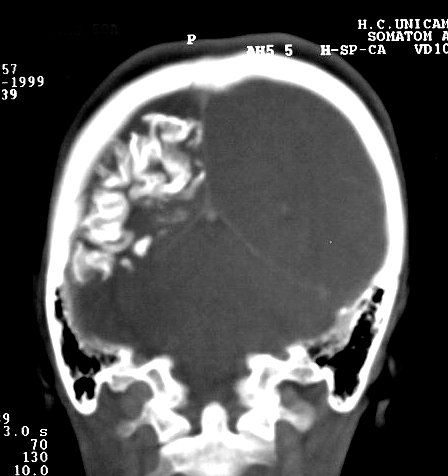
Com o tempo, desenvolve-se atrofia do hemisfério, mas é incomum haver atrofia na época do início das convulsões. A atrofia acompanha-se de gliose da substância branca, com hiposinal em T1 e hipersinal no TR longo. Há também aumento dos vasos periventriculares e subependimários, visualizável na TC e RM, em conseqüência da dilatação das veias da substância branca, o que por sua vez resulta de lentificação de fluxo ou trombose no sistema venoso superficial malformado. Por isso, as veias profundas da substância branca podem aparecer como pontos de flow void. Com a hemiatrofia cerebral desenvolve-se assimetria do crânio. Há espessamento da calota craniana ipsilateral, com aumento da espessura da díploe, e aumento de volume dos seios paranasais e das células da mastóide. Anomalias oculares
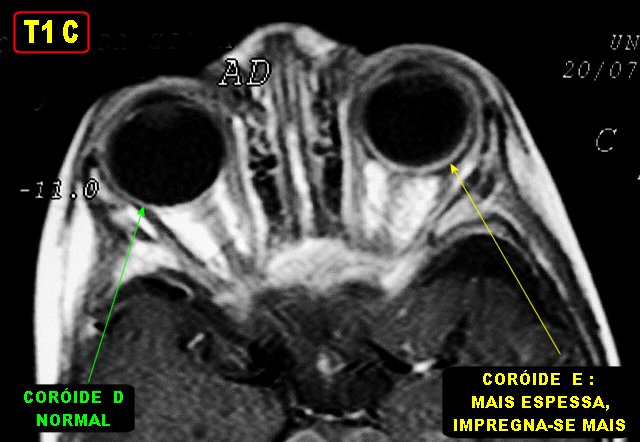
são vistas em 30% dos pacientes como angiomas da coróide
e esclera. Há glaucoma, com dor ocular ou retroorbitária,
ou descolamento da retina com deterioração visual rápida.
Se o glaucoma começa na vida intrauterina, o globo aumenta de volume
e fica um pouco alongado devido à pressão intraocular aumentada,
o que se conhece por buftalmia (olho de boi). O motivo do glaucoma é
a menor reabsorção dos fluidos intraoculares devido à
vascularização anormal da coróide.
O angioma da coróide correlaciona-se com doença bilateral e com a extensão do angioma facial, mas não há correlação com o tamanho do angioma pial. O angioma da coróide é observado em RM como espessamento da parede posterior do globo ocular, com impregnação por contraste, preferencialmente usando seqüências com supressão do sinal da gordura. Fora do sistema nervoso não há tipicamente anormalidades, mas pode haver angiomas de vísceras e extremidades. Os angiomas podem ser difusos ou localizados, podem ocorrer em intestinos, rins, baço, ovários, tiróide, pâncreas ou pulmões, o que é chamado de síndrome de Klippel-Trenaunay-Weber por alguns autores. Principais fontes:
|
||||||||||||||||||||||
|
|
|
|