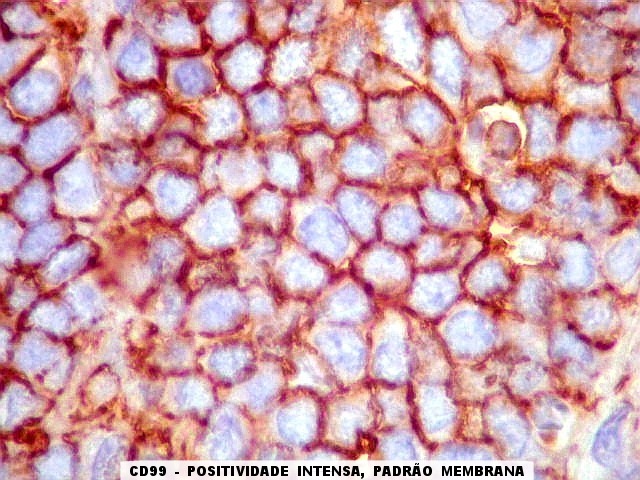
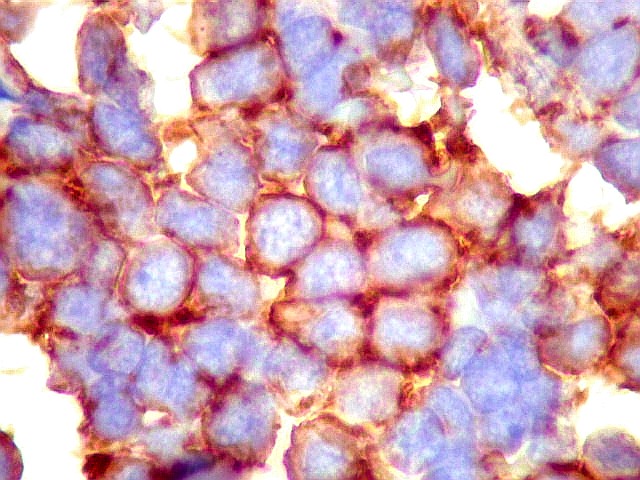
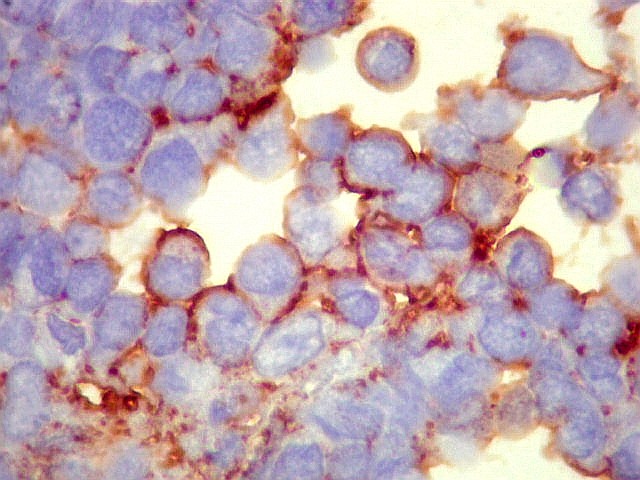
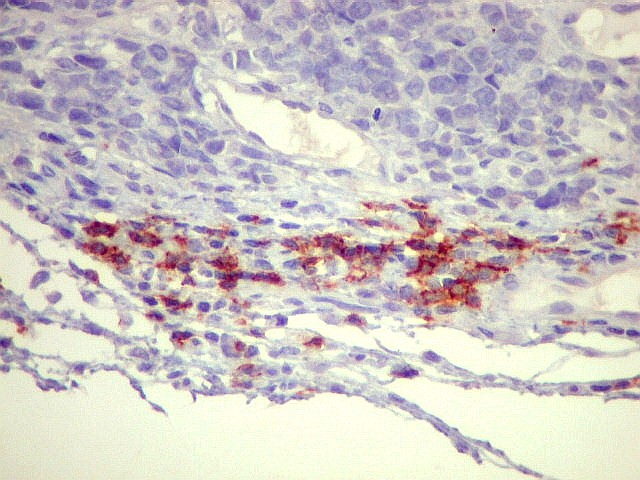
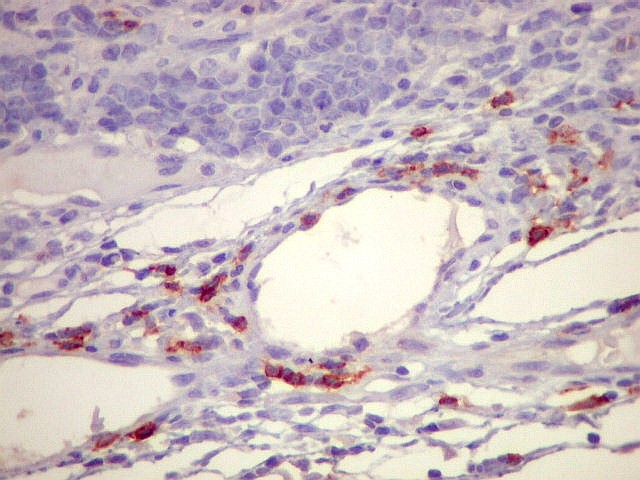
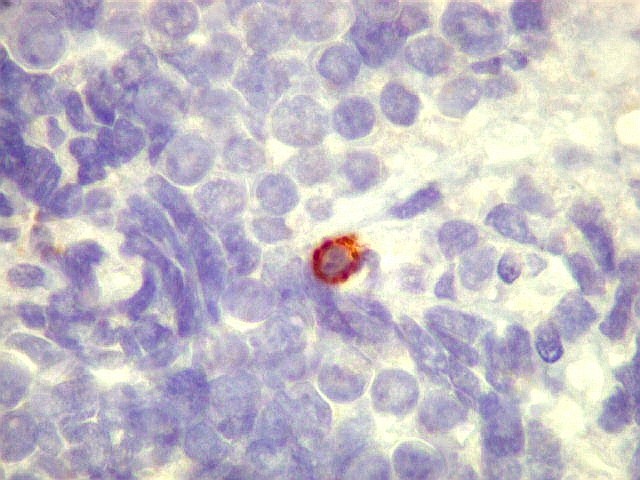
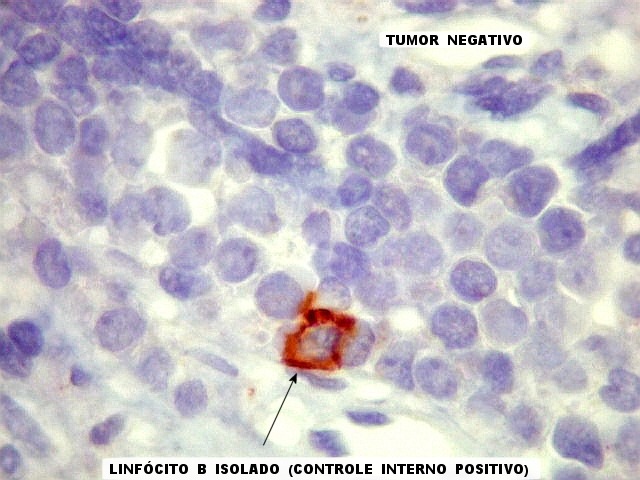
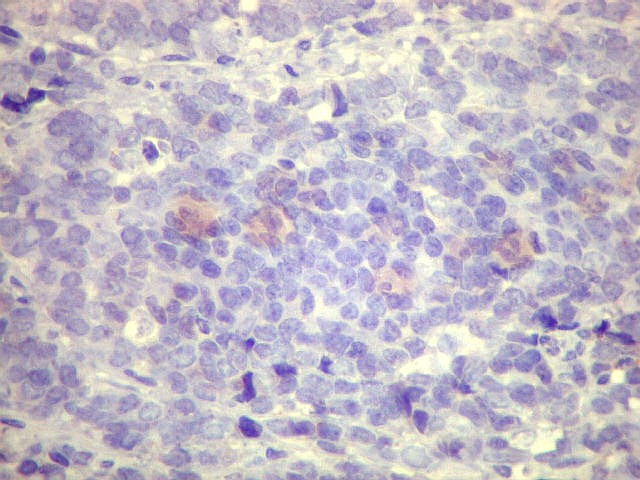
| Sarcoma
de Ewing e Tumor neuroectodérmico primitivo periférico (pPNET).
O sarcoma
de Ewing e o pPNET são tumores malignos de pequenas células
redondas primários de osso ou tecidos moles. Apresentam dificuldades
de diagnóstico diferencial por que as células redondas neoplásicas
são semelhantes às dos linfomas, rabdomiossarcomas, neuroblastomas
e carcinomas do tipo oat cell. O sarcoma de Ewing e o pPNET têm
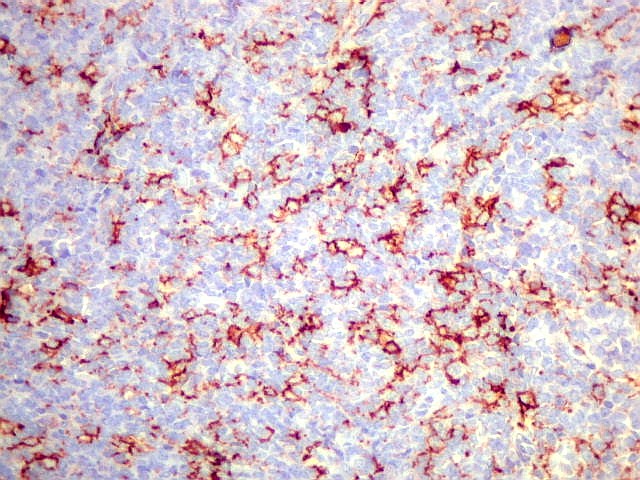
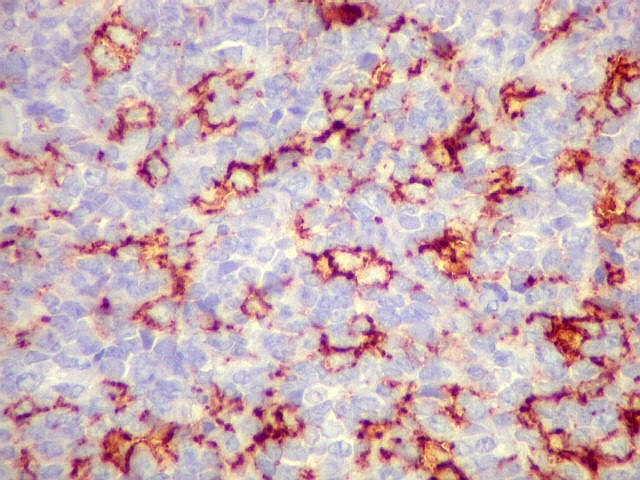
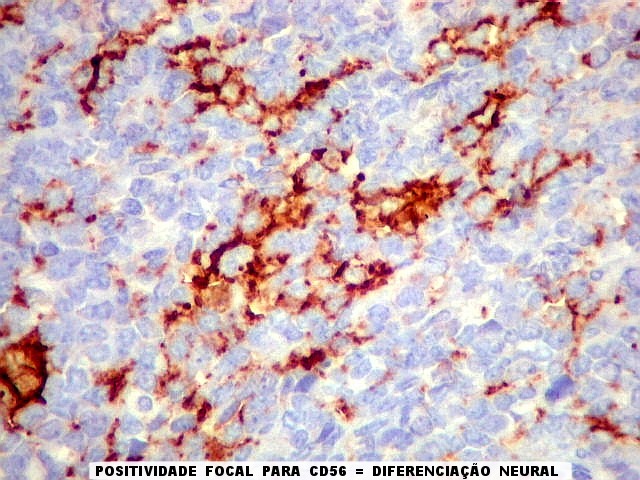
fenótipo neural e compartilham a mesma translocação
cromossômica. Por isso, devem ser vistos como o mesmo tumor, com
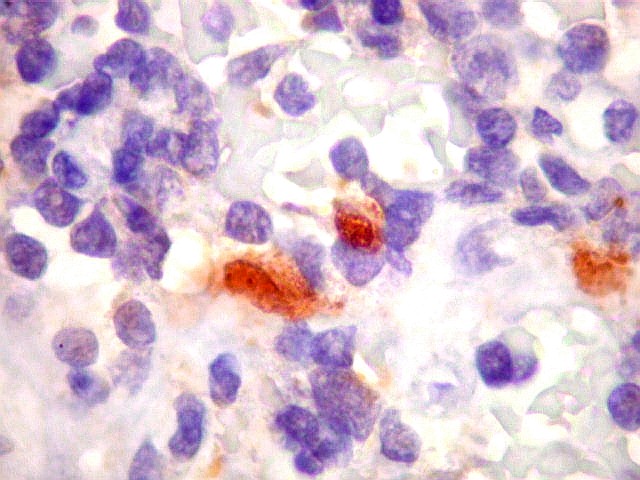
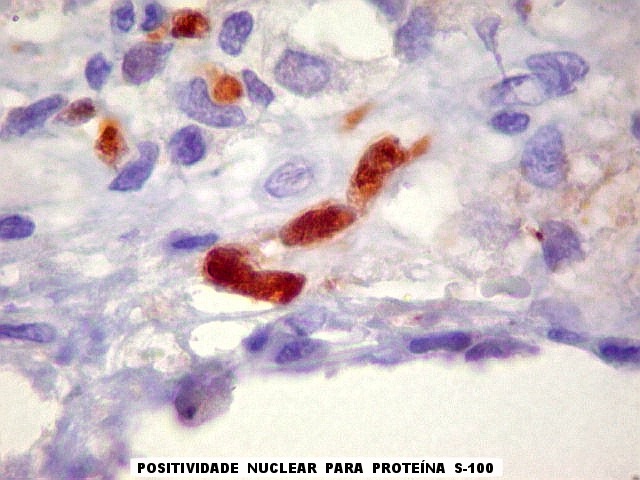
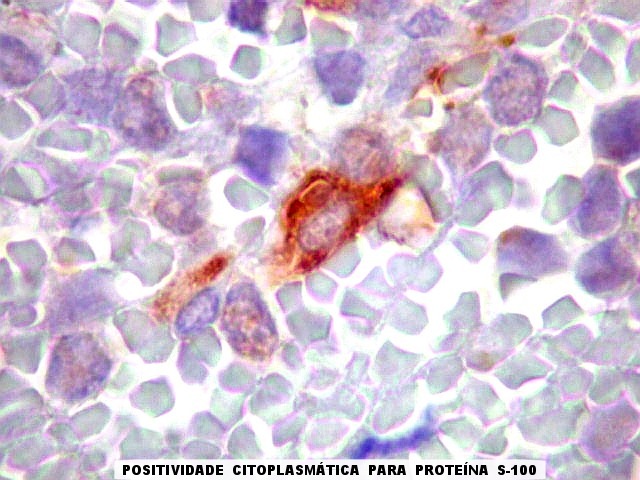
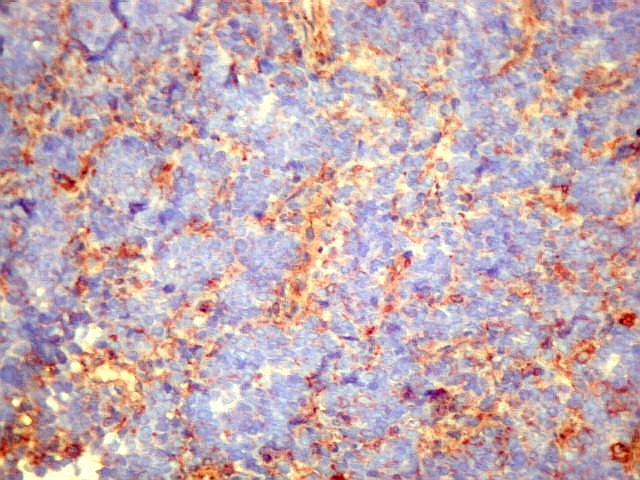
graus variáveis de diferenciação neural. Os tumores
que demonstram diferenciação neural em microscopia óptica,
imunohistoquímica e microscopia eletrônica são tradicionalmente
rotulados como pPNETs. Quando indiferenciados, são denominados sarcoma
de Ewing.
Incidência.
Os dois tumores correspondem a cerca de 6 a 10% dos tumores malignos primários
de osso e perdem apenas dos osteossarcomas no grupo dos sarcomas ósseos
em crianças. Dos sarcomas ósseos, o sarcoma de Ewing
tem a idade de apresentação mais precoce. A maioria dos pacientes
está na faixa de 10 a 15 anos e 80% ocorrem antes dos 20 anos.
Há discreta preferência pelo sexo masculino e forte predominância
em caucasianos (negros são raramente afetados).
Clínica.
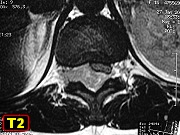
O local mais comum de origem é nas diáfises dos ossos tubulares
longos, especialmente o fêmur, e nos ossos chatos da pelve.
Apresentam-se como massas expansivas e dolorosas e podem se acompanhar
de sinais inflamatórios. Alguns pacientes podem ter sinais sistêmicos,
como febre, aumento da taxa de hemossedimentação, anemia
e leucocitose. Exames radiológicos mostram tumor destrutivo,
lítico, permeativo, que se estende aos tecidos moles próximos.
A reação periostal com deposição de camadas
de osso reativo lembrando bulbo de cebola é característica.
Atualmente o tratamento com quimioterapia permite sobrevida de 5 anos de
75% e curas em 50%.
Genética.
Em cerca de 85 % dos SE/pPNETs há uma translocação
recíproca entre cromossomos 11 e 22 [t(11;22)(q24;q12)]. Em 5-10%
outras translocações são encontradas. Em todos os
casos há fusão do gene EWS no cromossomo 22q12 a um membro
da família ETS de fatores de transcrição, especialmente
FLT1. O gene híbrido mais comum (EWS-FLI1), gerado a partir
da translocação 11-22, funciona como um oncogene dominante.
A proteína quimérica resultante atua como um fator de transcrição
intrinsecamente ativo, que estimula a proliferação celular.
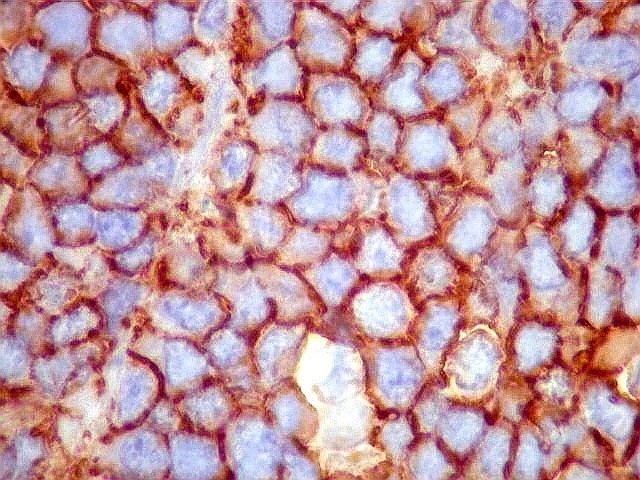
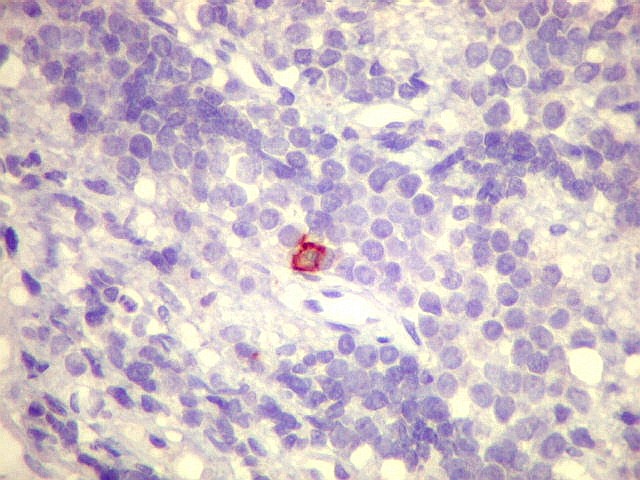
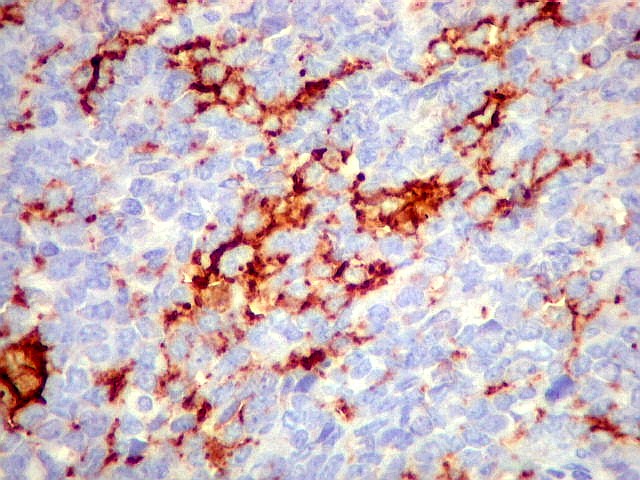
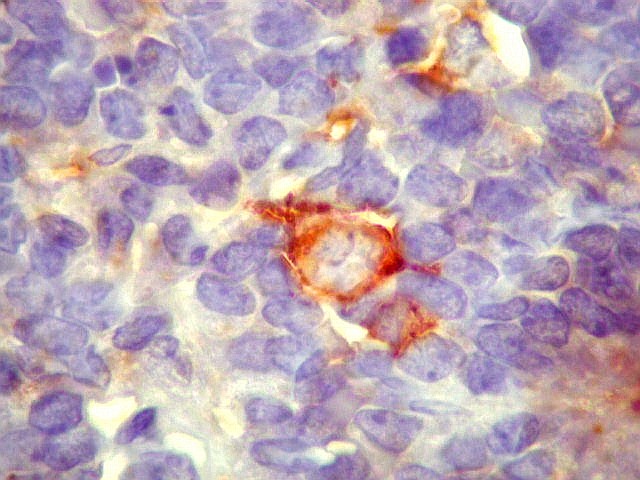
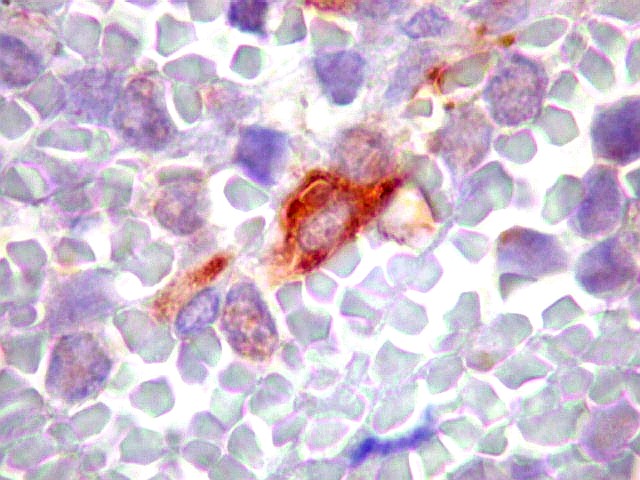
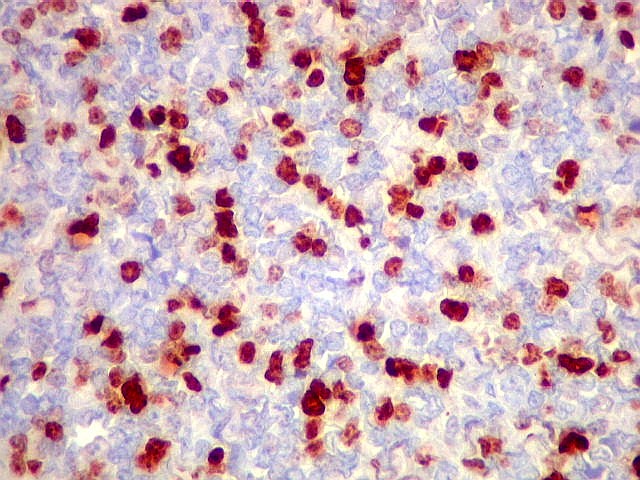
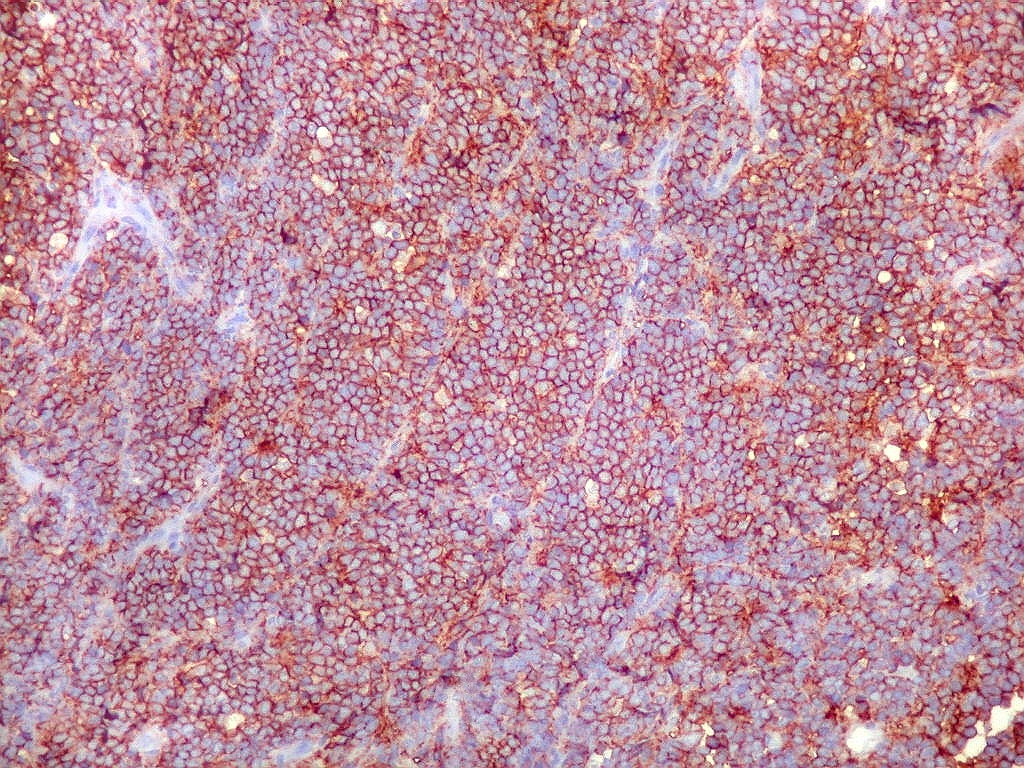
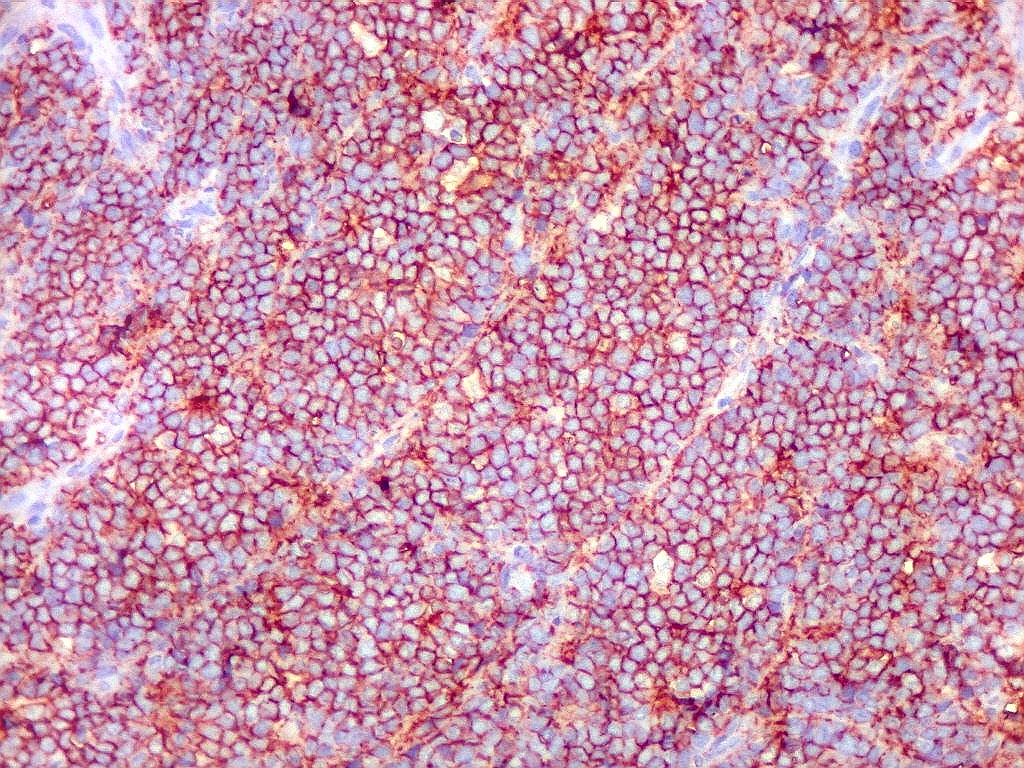
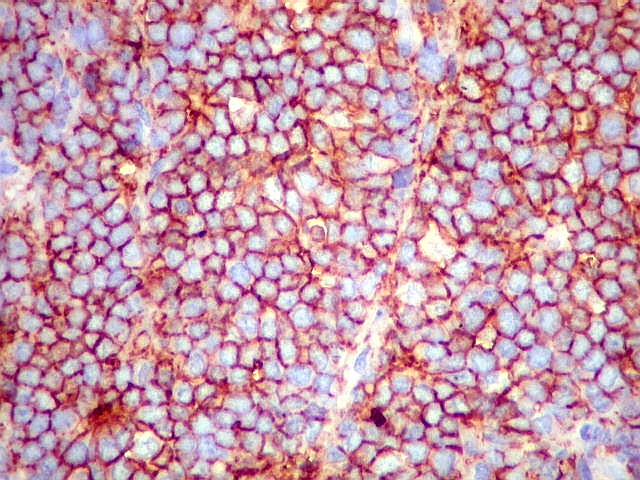
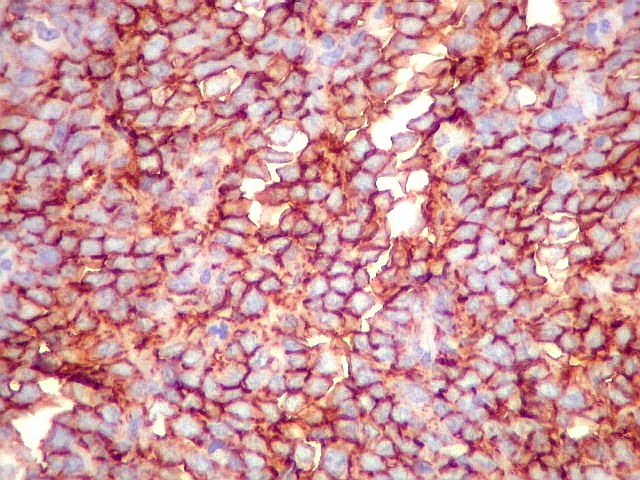
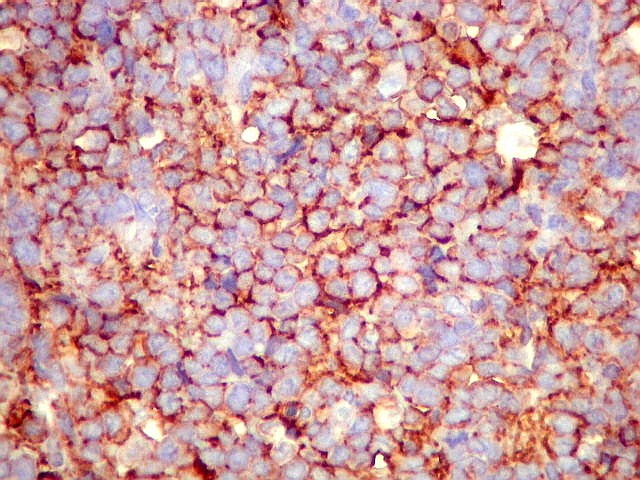
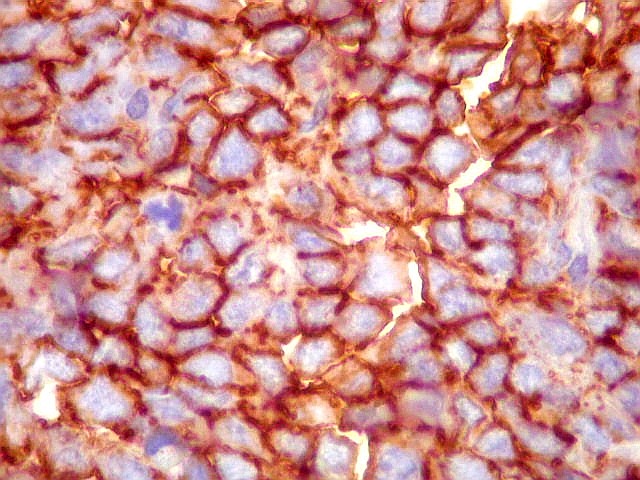
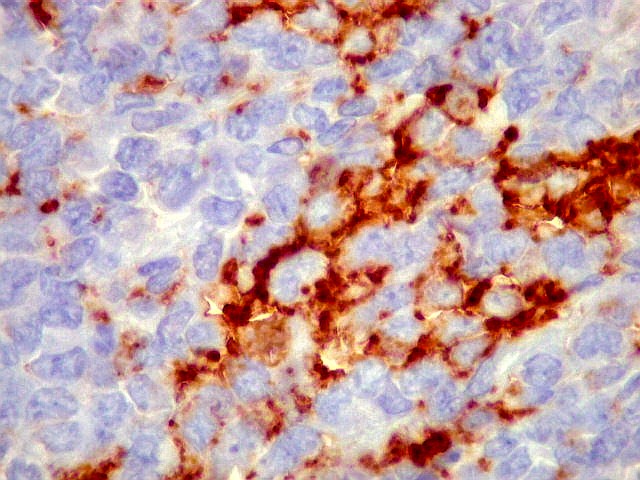
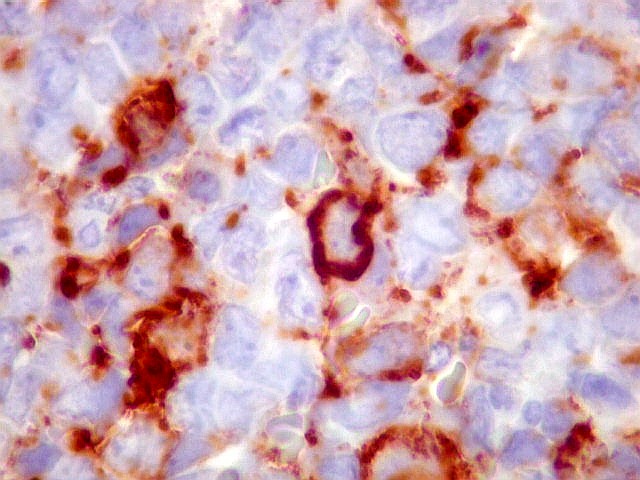
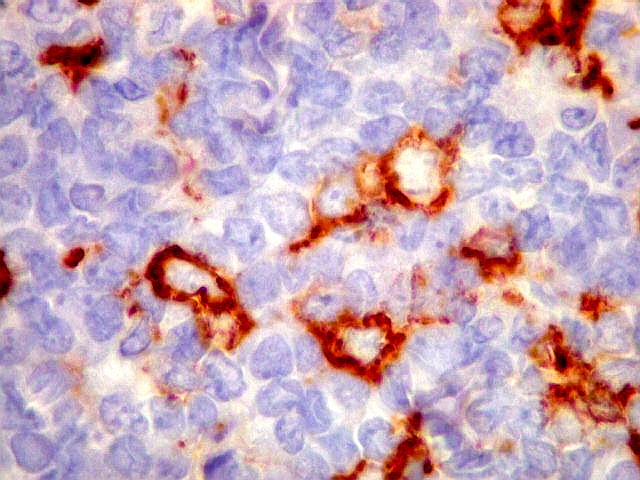
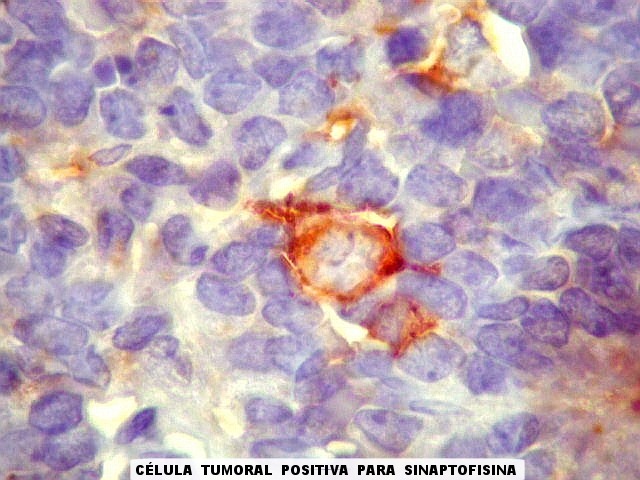
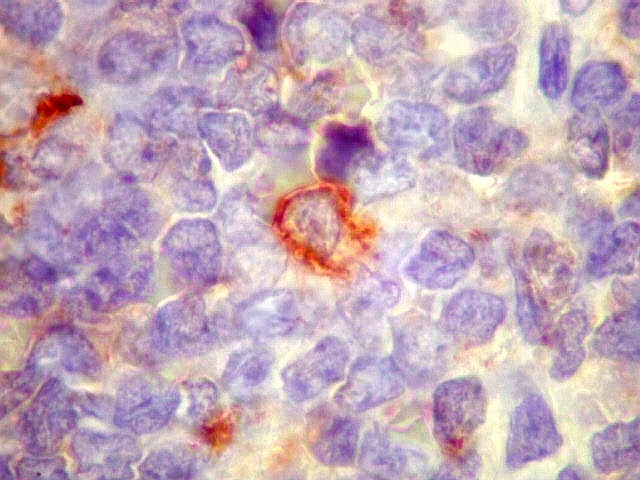
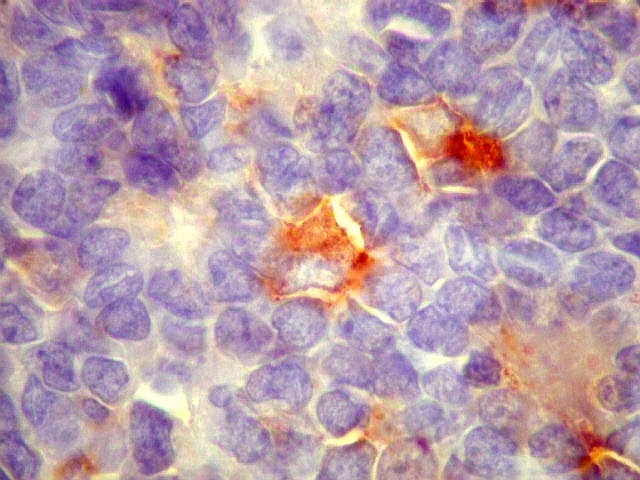
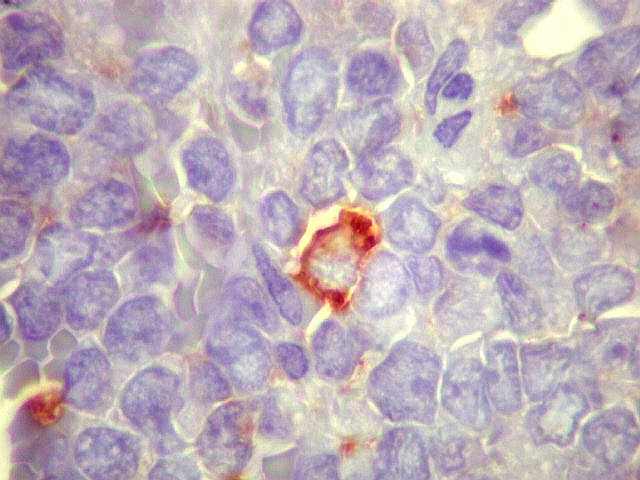
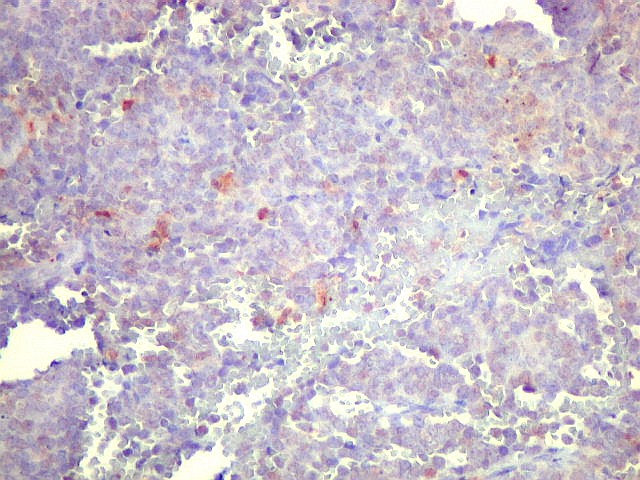
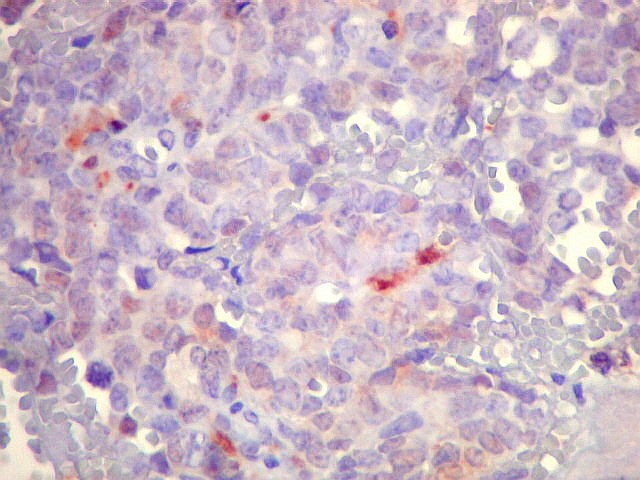
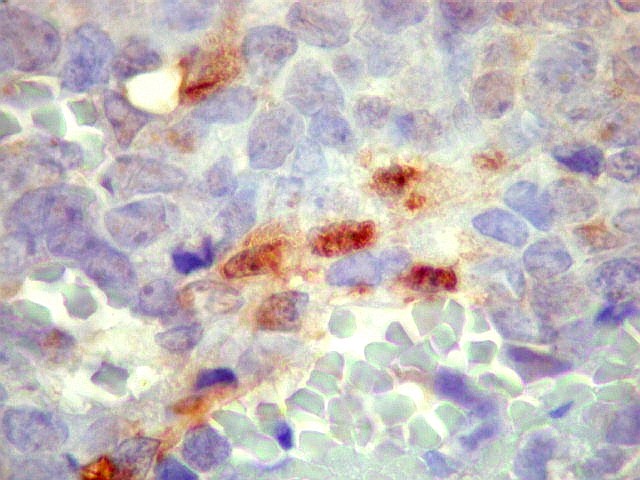
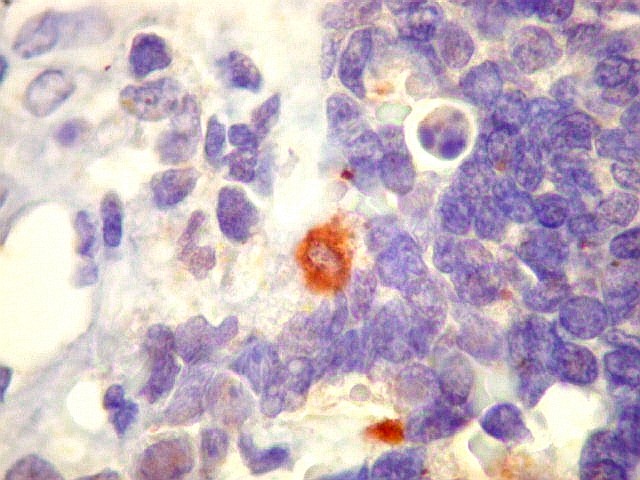
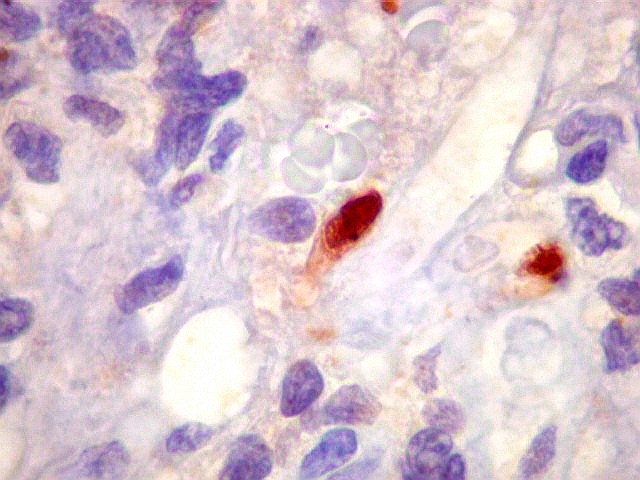
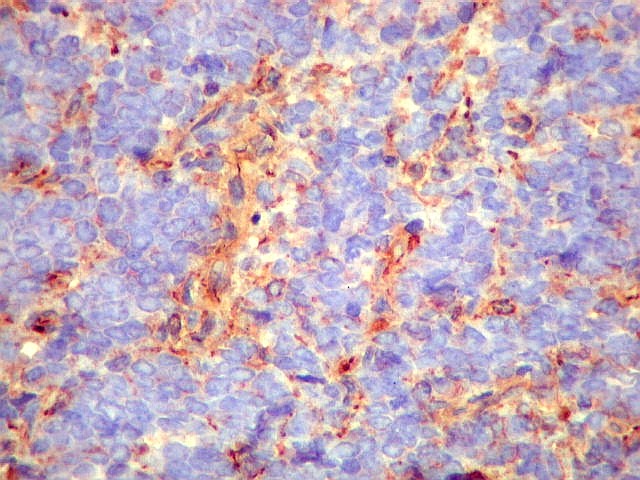
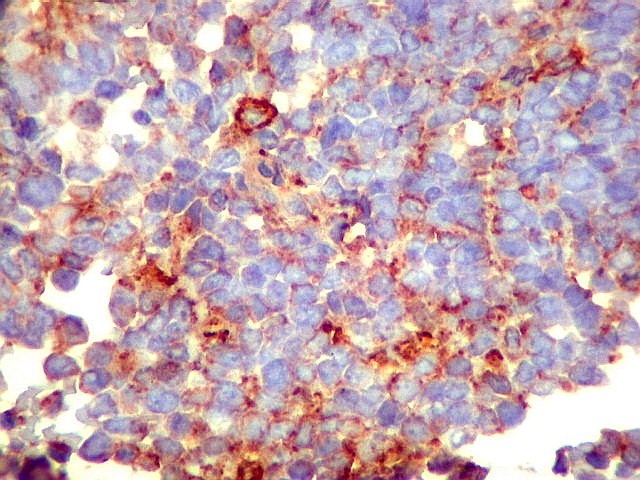
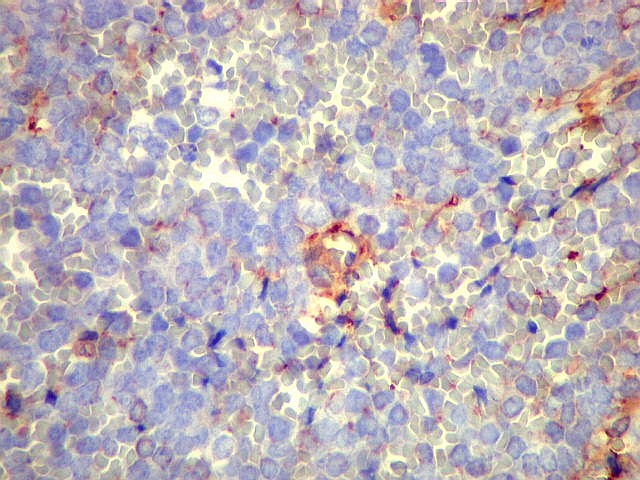
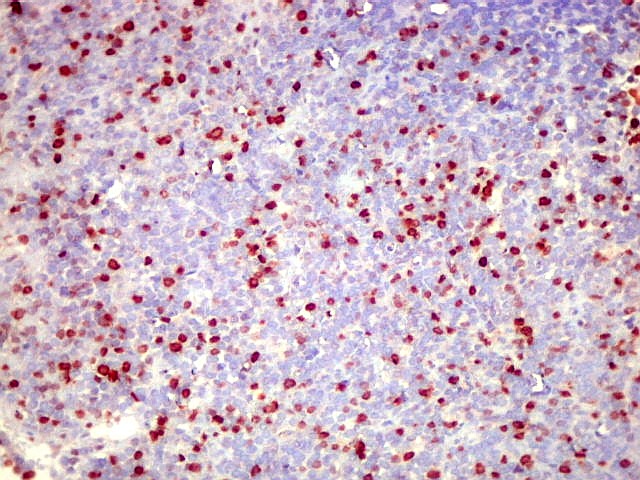
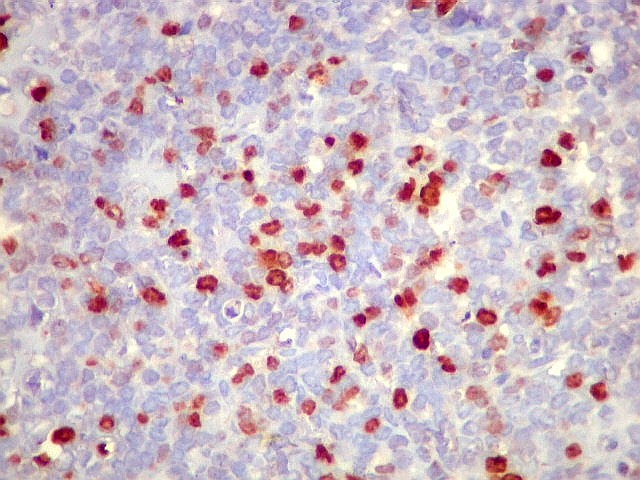
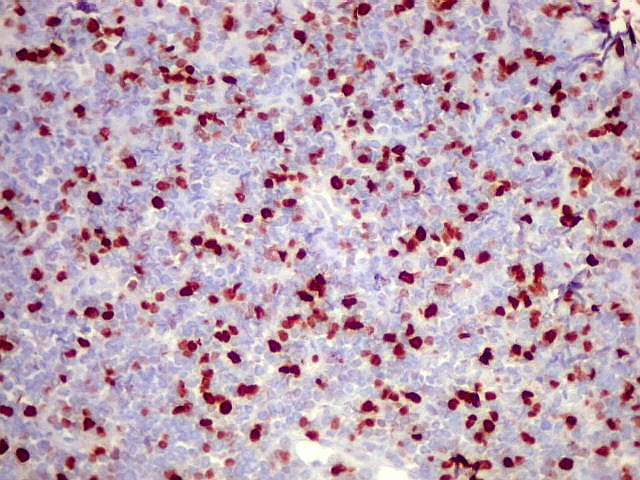
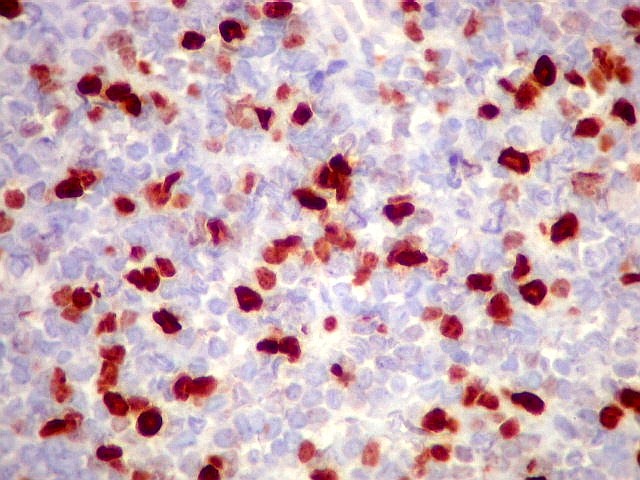
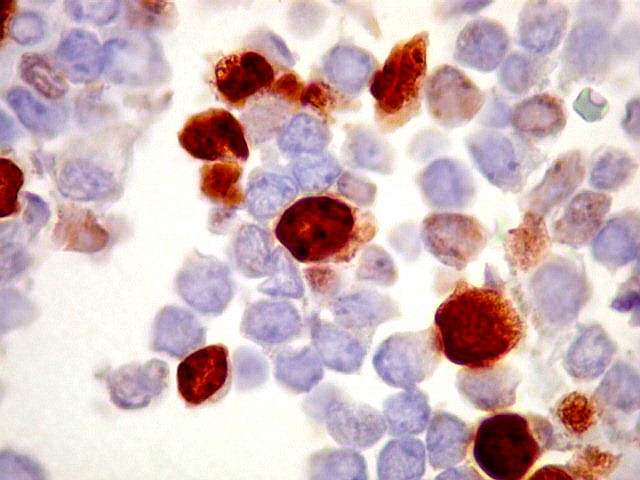
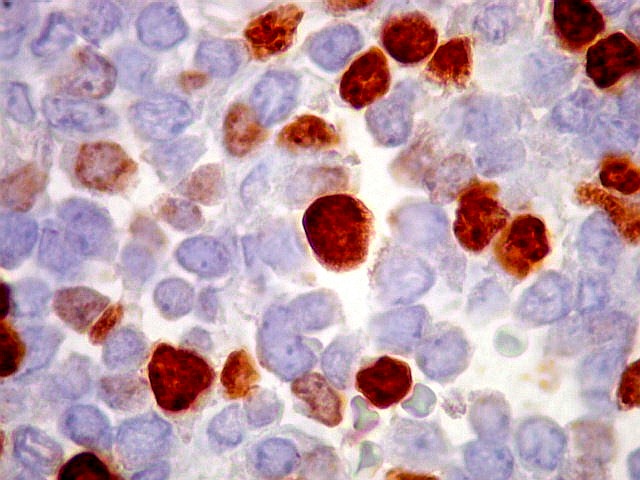
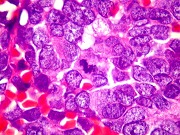
Morfologia.
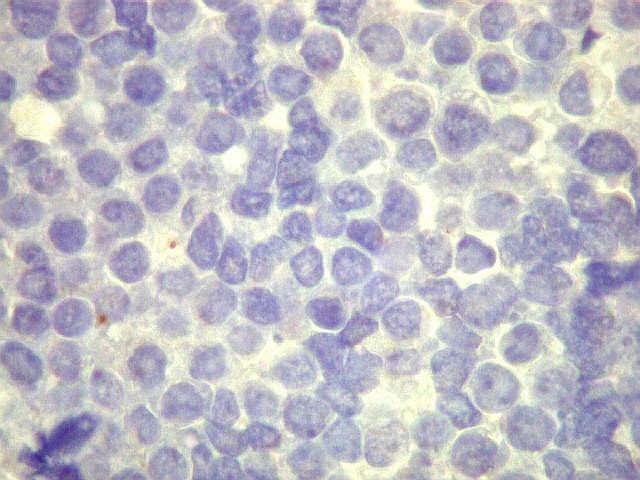
Os SE/pPNETs iniciam-se na medula óssea, de onde invadem o córtex
e o periósteo para produzir massa em tecidos moles. As células
uniformes, pequenas e redondas, em arranjo sólido, são pouco
maiores que linfócitos. O citoplasma escasso pode acumular glicogênio
e aparecer claro. Há pouco estroma, mas pode haver formação
de septos fibrosos. Há poucas mitoses em relação à
densa celularidade do tumor. Rosetas de Homer Wright, quando presentes,
indicam diferenciação neural.
Fonte.
Chapter 26 Bones, Joints and Soft Tissue Tumors. A. E. Rosenberg.
in Robbins and Cotran Pathologic Basis of Disease. 7th. Ed. Kumar V,
Abbas AK, Fausto N (editors). Elsevier, Saunders, 2005.
pp.1301-2. |