| Tumores
de pequenas células redondas.
Termo genérico
para um grupo de tumores malignos altamente agressivos compostos por células
indiferenciadas pequenas e monótonas com alta relação
núcleo-citoplasma.
Tumores
que incidem mais na idade pediátrica:
-
sarcoma de
Ewing,
-
neuroepitelioma
periférico, também conhecido como tumor neuroectodérmico
primitivo (PNET), ou sarcoma de Ewing extraesquelético,
-
neuroblastoma
periférico (tipo clássico),
-
rabdomiossarcoma,
-
tumor desmoplásico
de pequenas células redondas (DSRCT),
-
linfomas e
leucemias, geralmente do tipo linfoblástico,
-
osteossarcoma
de pequenas células e
-
condrossarcoma
mesenquimatoso.
Tumores de
que incidem mais em adultos:
-
carcinoma de
pequenas células (ou indiferenciado ou neuroendócrino),
-
neuroblastoma
do olfatório
-
carcinoma neuroendócrino
cutâneo ou de células de Merkel,
-
melanoma de
pequenas células
-
tumor extramedular
de células mielóides ou sarcoma granulocítico.
O diagnóstico
diferencial é dificultado pela semelhança morfológica
em HE, superposição entre as manifestações
clínicas, como localização e idade do paciente, alta
incidência de metástases, e apresentações atípicas
ou raras como neuroblastoma em adultos, rabdomiossarcoma cutâneo
primário, e carcinoma de células de Merkel linfonodal primário.
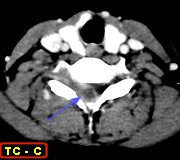
SARCOMA
DE EWING E PNET PERIFÉRICO.
Sarcoma
de Ewing (com exemplos esqueléticos e extraesqueléticos),
PNET
e tumor de Askin (PNET toracopulmonar) são um grupo de tumores
de pequenas células com feições clínicas e
patológicas parcialmente superponíveis, que se originam na
infância ou idade adulta jovem.
Classicamente,
o sarcoma de Ewing é um tumor primário de osso, enquanto
que o PNET usualmente se origina em tecidos moles. Contudo, sarcoma
de Ewing pode se apresentar fora do esqueleto e há casos raros de
PNET originado em osso. Isto sugere que SE/PNET seria só um tipo
de tumor com um espectro de apresentações, estando SE num
extremo e PNET no outro. Ambos tumores compartilham uma anormalidade genética
única, uma translocação t(11;22)(q24;q12), que foi
identificada em SE em 1983, em PNET em 1984 e no tumor de Askin em 1985.
SE é
considerado um tumor indiferenciado, enquanto que diferenciação
neural é característica dos PNETs. Contudo, evidência
de diferenciação neural é encontrada em alguns casos
de SE clássico, por IH, ME ou técnicas moleculares. Segundo
Enzinger e Weiss o tumor deve ser diagnosticado como PNET se mostrar rosetas
de Homer Wright ou de Flexner-Wintersteiner bem definidas, imunorreatividade
para 2 ou mais marcadores neurais ou evidência ultraestrutural de
diferenciação neural.
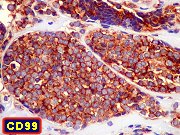
Padrão imunohistoquímico
em SE/PNET
CD99
Anticorpos monoclonais para CD99 (MIC2) reconhecem a glicoproteína
de superfície celular que é produto do gene de fusão
EWS/FLI-1 criado pela translocação. Têm sensibilidade
> 95% para SE ou PNET. Neuroblastomas periféricos ou clássicos,
e os neuroblastomas do olfatório são negativos para CD99.
FLI-1 = Friend leukemia virus integration 1.
Infelizmente,
CD99 não é específico para SE/PNET, sendo reativo
em vários outros tumores, inclusive:
-
linfomas linfoblásticos
(57 a 100%), que devem ser excluídos com LCA, CD3, CD20, TdT.
-
Outros linfomas
Hodgkin e não Hodgkin podem às vezes reagir com CD99.
-
tumores extramedulares
de células mielóides (sarcoma granulocítico) (diferenciar
com mieloperoxidase (MPO ou MPX) e CD43).
-
rabdomiossarcoma
embrionário ou alveolar (diferenciar com actina específica
de músculo (MSA) e desmina). Estes marcadores por sua vez também
não são específicos, podendo reagir em até
17% de tumores não miogênicos de tecidos moles, presumivelmente
reagindo com o componente miofibroblástico. Marcadores altamente
sensíveis e específicos para músculo esquelético
são miogenina e myoD1 e estes não mostraram reatividade em
SE/PNET.
-
tumor desmoplásico
de pequenas células redondas (6/17 casos em um estudo),
-
e outros.
SE/PNET
podem expressar citoqueratinas, mas não coexpressam citoqueratinas
e desmina, enquanto que esta co-expressão é altamente característica
do chamado tumor desmoplásico de pequenas células redondas
(DSRCT). Há também importantes diferenças clínicas,
com o SE/PNET aparecendo como massas esqueléticas ou em tecidos
moles, enquanto o DSRCT manifesta-se como envolvimento seroso abdominal
disseminado. Já foi também relatado em pleura, ovário,
escroto e cérebro.
Imunorreatividade
para NCAM (neural cell adhesion molecule ou CD56) está tipicamente
presente em neuroblastomas periféricos e ausente em SE/PNET. Já
beta2-microglobulina
se comporta de forma oposta. Em neuroblastomas periféricos e do
olfatório há células dendríticas que circundam
os lóbulos tumorais e são reativas para S100. SE/PNET
não mostra este padrão.
NEUROBLASTOMA
PERIFÉRICO OU CLÁSSICO.
São
geralmente vistos em crianças pequenas, com até 50% até
os 2 anos de idade. São notados pela habilidade em sofrer maturação
ou regressão espontâneas. Originam-se no sistema nervoso simpático,
com sítios primários na medula da supra-renal, retroperitônio,
tórax, cabeça, pescoço e pelve. Alta percentagem de
casos já se apresenta com metástases ósseas, hepáticas,
linfonodais ou cutâneas.
As duas
anormalidades
genéticas mais comuns são perda de heterozigose para
o braço curto do cromossomo 1 e amplificação do oncogene
N-myc.
O principal
diagnóstico diferencial é com SE/PNET. A presença
de CD99 virtualmente exclui neuroblastoma. Em um estudo recente, 105
neuroblastomas foram negativos. Ao contrário, imunorreatividade
para NCAM (neural cell adhesion molecule ou CD56) está tipicamente
presente em neuroblastomas periféricos e ausente em SE/PNET. Já
beta2-microglobulina se comporta de forma oposta. Exemplos : um estudo
encontrou NCAM em 16/16 neuroblastomas e 3/33 SE/PNET. Outro - beta2-microglobulina
em 13/17 SE/PNET e 0/26 neuroblastomas.
TUMOR DESMOPLÁSICO
DE PEQUENAS CÉLULAS REDONDAS.
Os primeiros
relatos de DSRCT intraabdominal são de 1989. Ocorrem mais em crianças
e adultos jovens com forte predominância masculina. A maioria tem
envolvimento peritonial difuso. Em 1992 foi identificada uma translocação
característica t(11;22)(p13;q12), (semelhante à t(11;22)(q24;q12),
do SE/PNET), que resulta em fusão dos genes EWS e WT1 (Wilms Tumor).
Antes da
identificação da fusão, os DSRCT eram definidos primariamente
pelo seu padrão IH característico , com coexpressão
de antígenos epiteliais (citoqueratinas, EMA), mesenquimais
(vimentina, desmina) e neurais (NSE). Em vários casos, reatividade
para desmina, vimentina ou queratina mostravam um padrão paranuclear
globular ou puntiforme (padrão dot), correspondendo
ultraestruturalmente a agregados paranucleares de filamentos intermediários.
DSRCT podem
expressar CD99 e alguns SE/PNET podem expressar citoqueratina ou desmina,
mas não coexpressam ambas. SE/PNET geralmente se apresentam como
lesões sólidas, enquanto DSRCT envolvem superfícies
serosas difusamente.
CARCINOMAS
DE PEQUENAS CÉLULAS.
Carcinomas
de pequenas células do pulmão, e os extrapulmonares
(muito menos freqüentes) são os tumores de pequenas células
redondas mais comuns que se originam em adultos. São tipicamente
muito agressivos e metastatizantes.
Usualmente
mostram reatividade para citoqueratinas e vários marcadores
neuroendócrinos, como NSE, sinaptofisina, cromogranina A, Leu-7
(CD57) e protein gene product (PGP) 9.5.
Os que reagem
para queratinas, mas não para marcadores neuroendócrinos,
são denominados carcinomas indiferenciados de pequenas células.
Os que reagem
para ambos são designados carcinomas neuroendócrinos de
pequenas células. Geralmente carcinomas de pequenas células
não reagem para CK20, enquanto isto ocorre no carcinoma de células
de Merkel. Há imunoreatividade dos carcinomas de pequenas células
pulmonares para TTF-1 (thyroid transcription factor 1), o que não
ocorre no carcinoma de células de Merkel.
O diagnóstico
diferencial de carcinomas de pequenas células inclui SE/PNET,
linfomas e neuroblastoma do olfatório.
Os carcinomas
de pequenas células tipicamente originam-se no pulmão
ou outras vísceras e não mostram reatividade para
CD99. SE/PNET originam-se em osso ou tecidos moles e mostram
reatividade para CD99. Contudo, pode haver superposição
entre carcinomas de pequenas células e SE/PNET a nível histológico,
imunohistoquímico e ultraestrutural e a distinção
entre eles nem sempre é clara.
Já
os carcinomas de pequenas células e os linfomas têm
perfis IH bem diferentes. LCA é positivo na maioria dos linfomas
e negativo nos CPC. Linfomas tipicamente não reagem para citoqueratinas
ou marcadores neuroendócrinos.
NEUROBLASTOMA
DO OLFATÓRIO.
Ocorre em
adultos na cavidade nasal alta ou seios nasais. Raros na infância.
São agressivos, recidivantes e metastatizantes, inclusive para o
pulmão. Não apresentam a translocação característica
do SE/PNET. São negativos para CD99, o que reforça que não
são tumores aparentados com SE/PNET.
O diagnóstico
diferencial do neuroblastoma do olfatório inclui SE/PNET e carcinomas
de pequenas células metastáticos. Usa-se CD99 para separar
neuroblastoma do olfatório de SE/PNET. Também neuroblastoma
do olfatório tem células dendríticas reativas para
S-100 ao redor de lóbulos de células tumorais, o que não
ocorre no SE/PNET nem nos carcinomas de pequenas células pulmonares
metastáticos. Estes últimos são positivos para queratinas,
enquanto neuroblastoma do olfatório é negativo ou só
focalmente positivo.
CARCINOMA
NEUROENDÓCRINO CUTÂNEO OU DE CÉLULAS DE MERKEL.
São
tumores de idosos, raros em jovens. Podem ocorrer na cabeça, pescoço,
tronco e extremidades. Tipicamente envolvem a derme. São agressivos,
com recidiva local freqüente (30%), metástases a linfonodos
regionais em 65% e distantes em 40%. Mesmo assim, são bem menos
agressivos que os carcinomas de pequenas células pulmonares ou extrapulmonares.
Por isso, é importante distingui-los.
A maioria
dos carcinomas de células de Merkel mostra reatividade paranuclear
em padrão dot para citoqueratinas, que corresponde
a agregados de filamentos intermediários. Contudo, o mesmo padrão
dot
pode ocorrer em carcinomas de pequenas células pulmonares ou extrapulmonares
metastáticos, e em vários outros tipos tumorais, particularmente
os que expressam 2 ou mais tipos de filamentos intermediários.
Reatividade
para CK20 mostrou utilidade diagnóstica, sendo positiva na
maioria dos carcinomas de células de Merkel (33/34 casos), freqüentemente
em padrão dot, e não é usual em carcinomas
de pequenas células pulmonares ou não (5/89 casos). Em contraposição,
TTF-1,
um fator de transcrição expressado seletivamente em tiróide,
pulmão e diencéfalo, foi encontrado em 35/36 carcinomas de
pequenas células pulmonares e em 0/21 carcinomas de células
de Merkel.
Diagnóstico
diferencial entre carcinomas de células de Merkel e linfomas
é feito com LCA e anti CKs. Contra melanoma amelanótico
de pequenas células, estes mostram reatividade para S-100 e
HMB-45, o que não ocorre com carcinomas de células de Merkel.
TUMOR EXTRAMEDULAR
DE CÉLULAS MIELÓIDES OU SARCOMA GRANULOCÍTICO.
Pode envolver
qualquer sítio e qualquer idade. A maioria dos pacientes têm
leucemia mielóide ou síndrome mieloproliferativa mas isto
pode faltar em 30% dos casos. Na grande maioria dos tumores há diferenciação
granulocítica. Num estudo de 28 casos, houve reatividade para CD43,
MPO e LCA em 100, 93 e 75%. CD43 indicador sensível de linhagens
aberrantes de células B.
Extraído de
-
Devoe K, Weidner
N. - Immunohistochemistry of small round-cell tumors. Seminars in
Diagnostic Pathology 17 (3):216-24, 2000.
|