|
|
1. Macro, HE |
|
|
|
1. Macro, HE |
|
| Masc. 19 a. Clique para história clínica, radiografias simples, tomografia computadorizada, ressonância magnética, angiografia digital, biópsia : macro, destaques; HE, tricrômico de Masson, reticulina, reticulina + safranina, imunohistoquímica (destaques) para VIM, 1A4, HHF-35, CD56, S-100, CD34, Ki-67, p53, microscopia eletrônica. Textos sobre fibroma condromixóide, fibronectina, integrinas. |
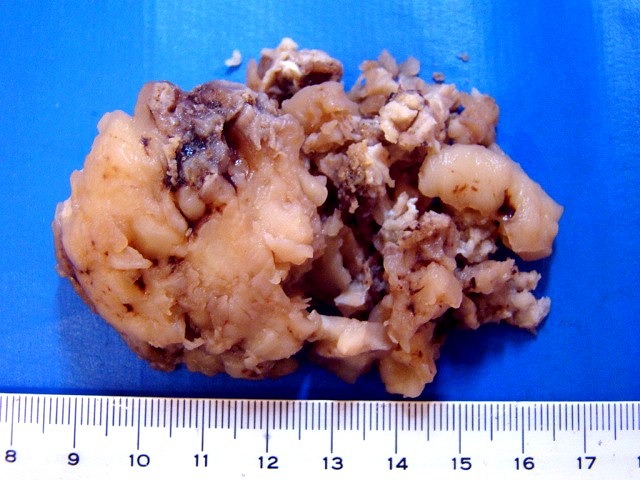
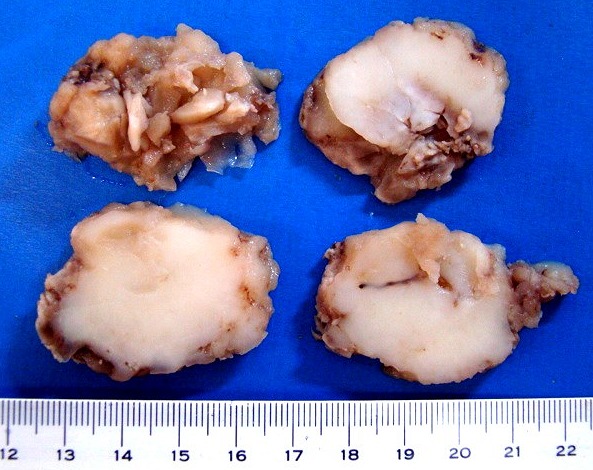
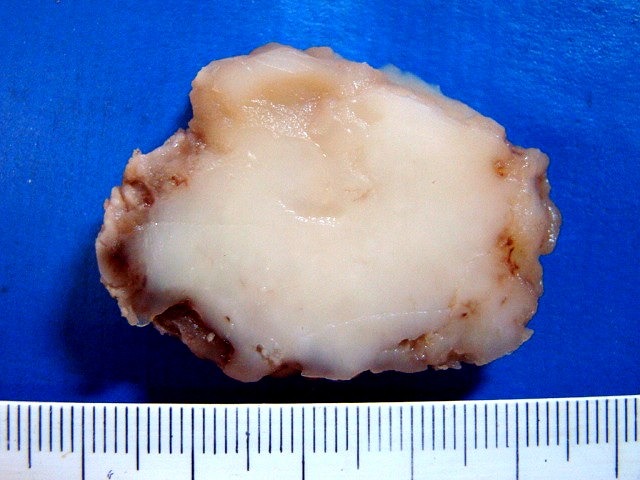
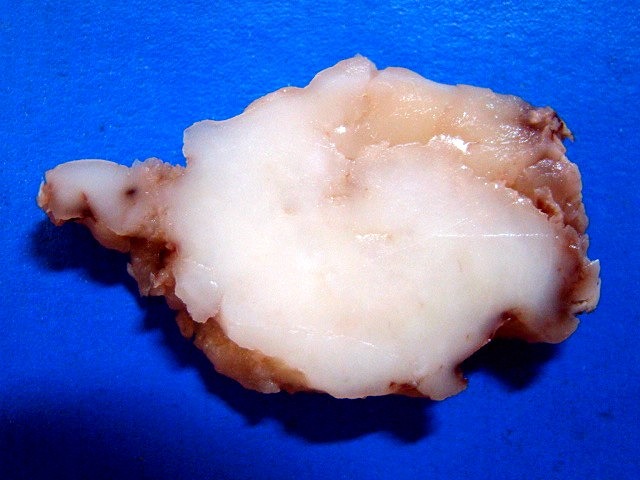
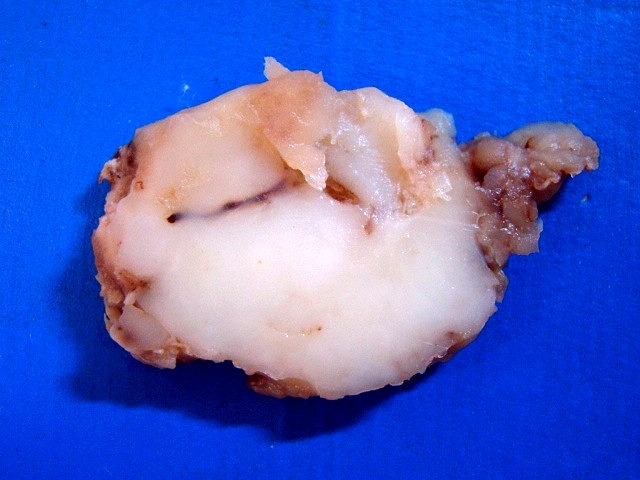
| Espécime. Massa de aspecto neoplásico medindo cerca de 7 x 4 x 4 cm, acinzentada, consistência firme e elástica, superfície externa lisa ou lobulada. Ao corte, cor branca e aspecto homogêneo. |
 |
 |
 |
 |
 |
 |
| Destaques da microscopia. | ||
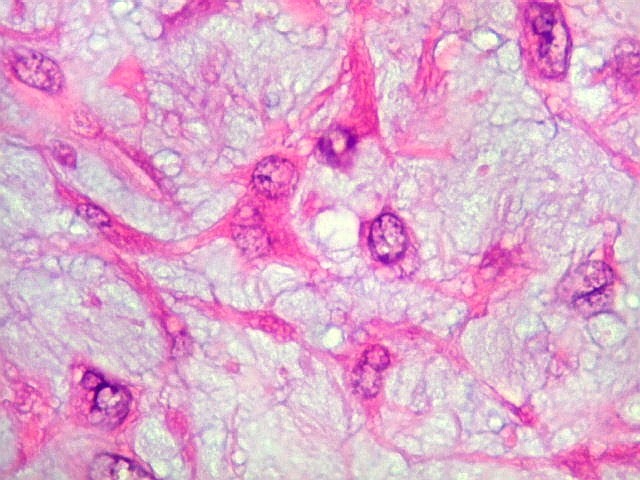
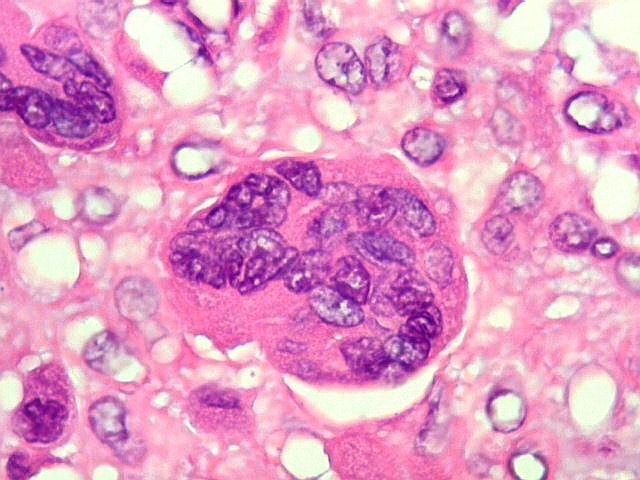
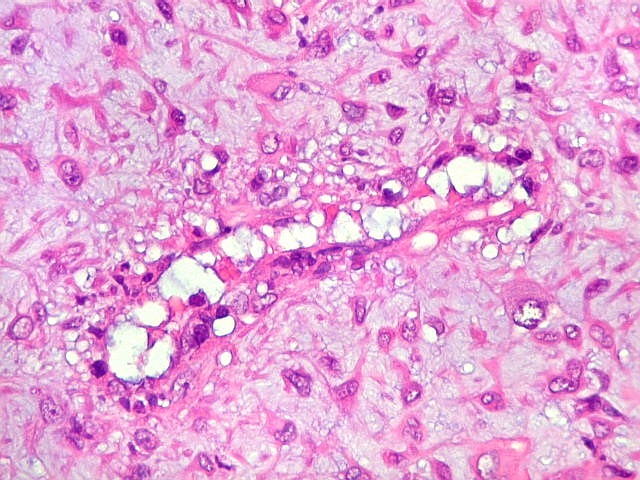
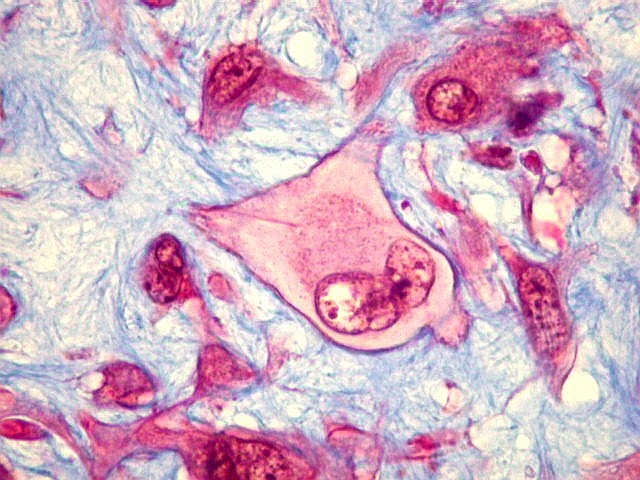
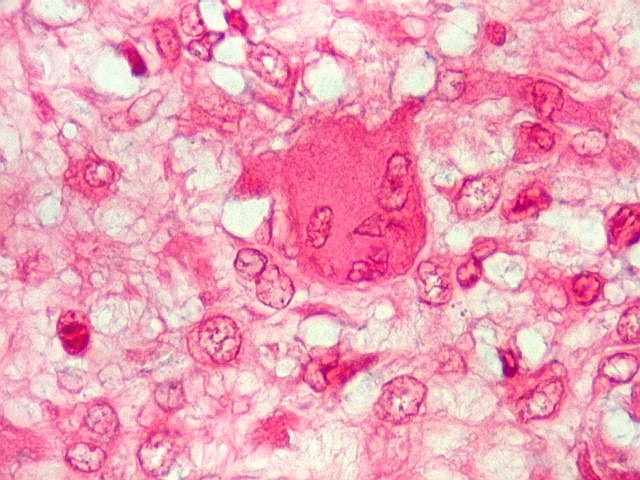
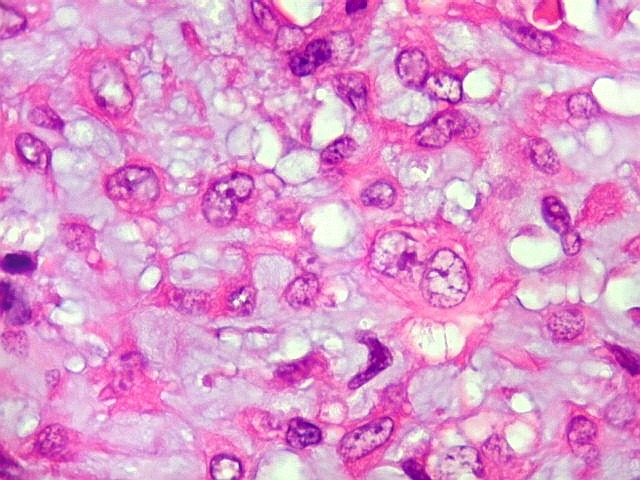
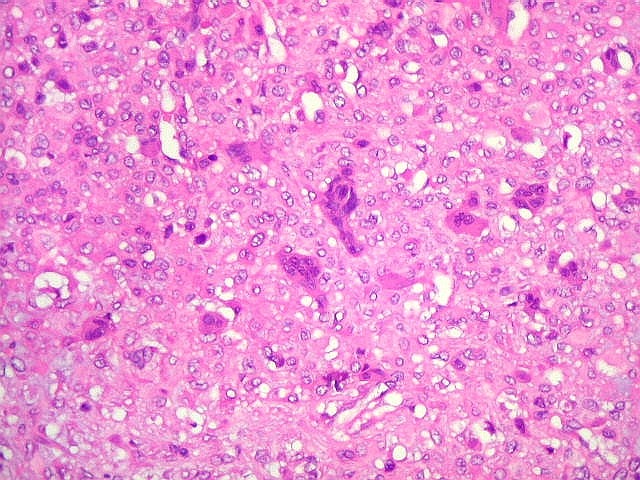
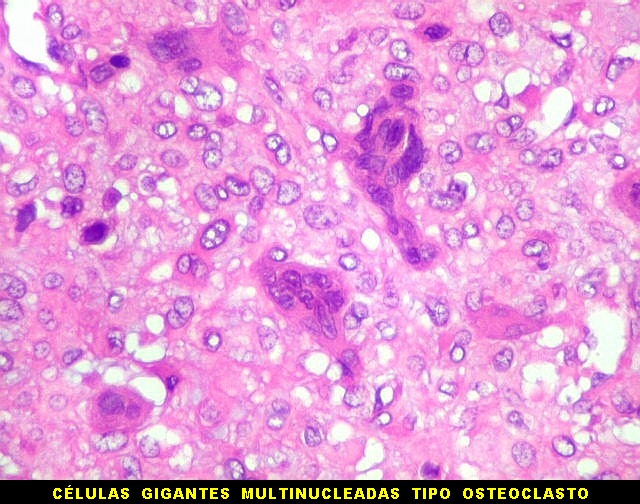
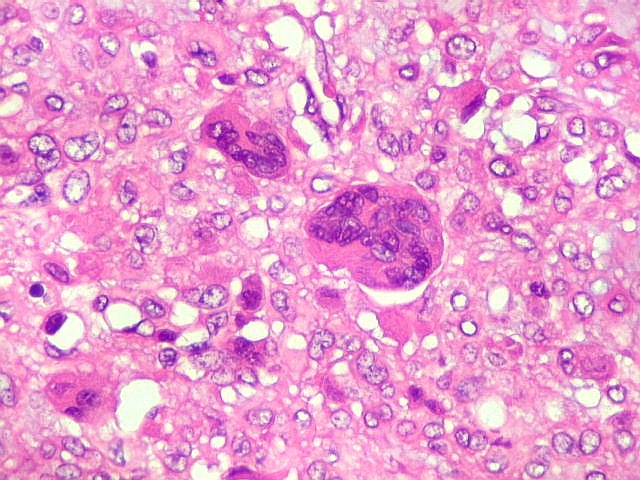
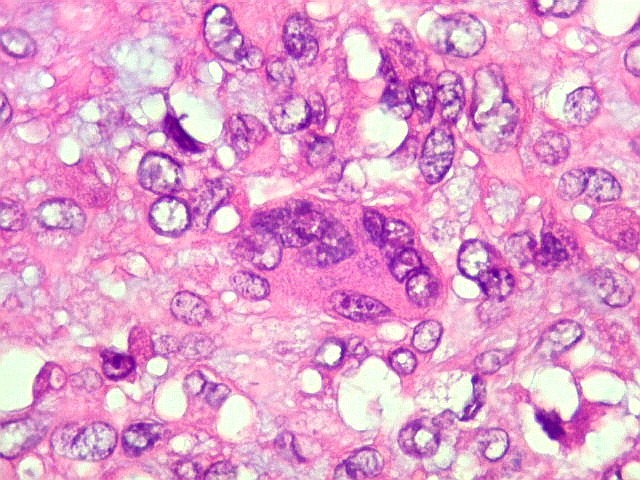
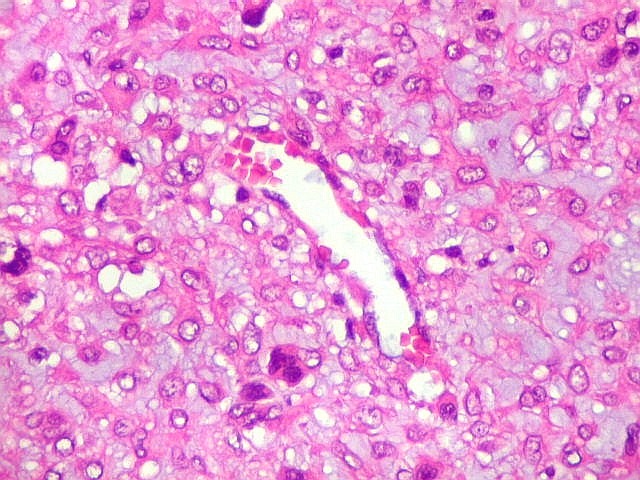
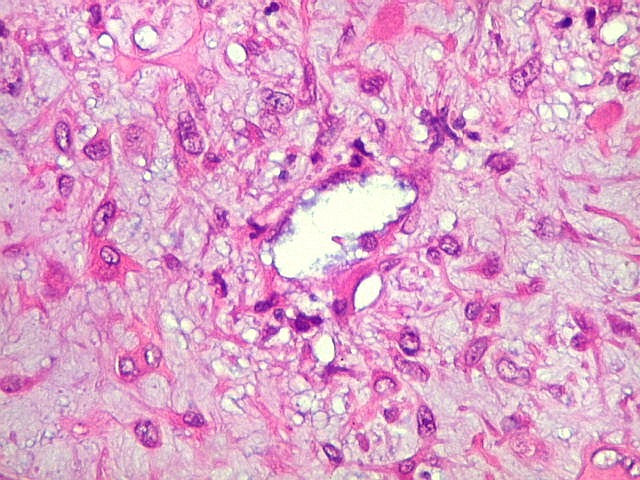
| HE. Células pleomórficas estreladas em abundante matriz mixóide | Células gigantes multinucleadas tipo osteoclasto esparsas, concentradas entre pseudolóbulos tumorais | Vasos : numerosos, finos, parede delgada |
 |
 |
 |
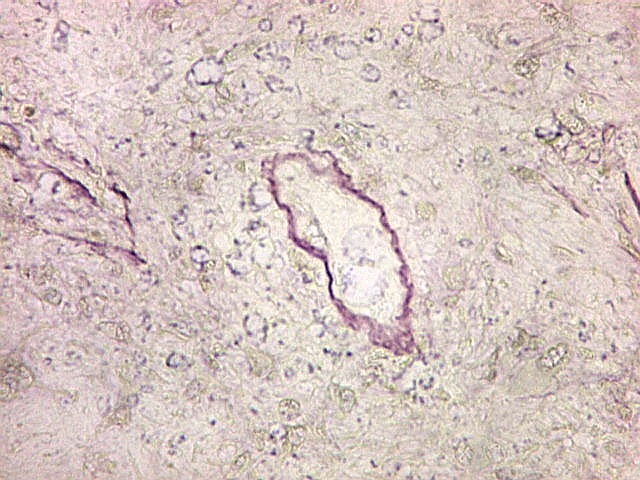
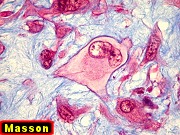
| Masson. Células de limites nítidos destacando-se contra a matriz mixóide. Nesta, fibrilas azuis, presumivelmente de colágeno | Reticulina. Restrita a vasos e a áreas com células fusiformes. Ausente entre células tumorais | Reticulina + safranina. Ausência de fibras reticulínicas entre as células neoplásicas ou osteoclastos |
 |
 |
 |
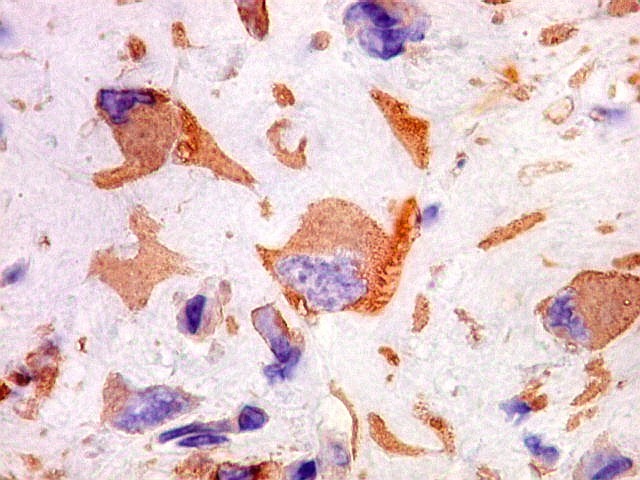
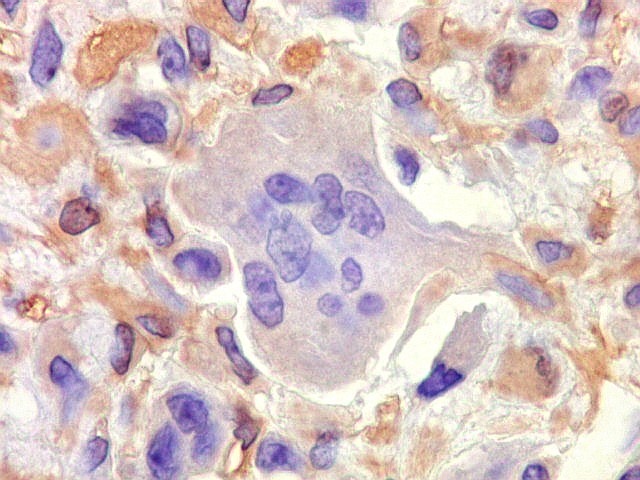
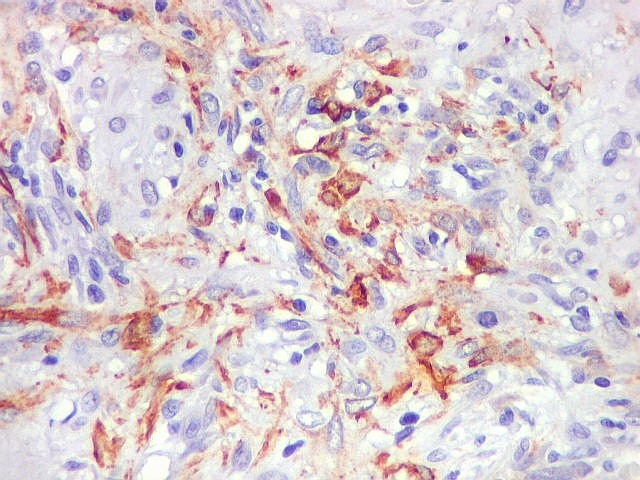
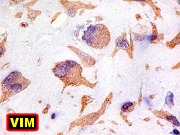
| VIM. Positividade citoplasmática forte e difusa nas células neoplásicas | Negativa nas células gigantes multinucleadas tipo osteoclasto | 1A4. Positividade citoplasmática focal em células neoplásicas, na maioria perivasculares. Positivo mais forte em áreas com células fusiformes. |
 |
 |
 |
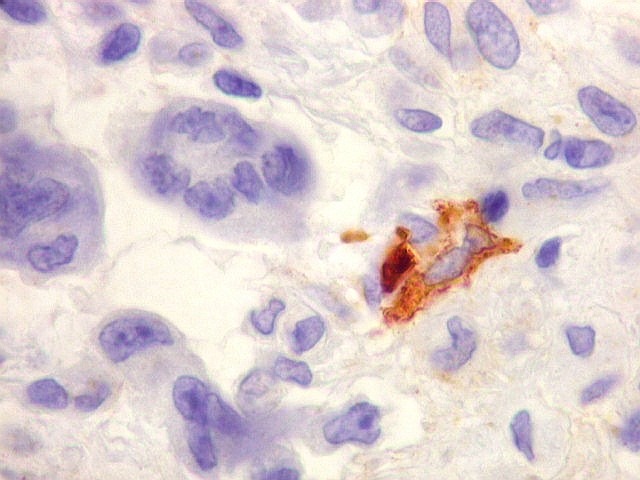
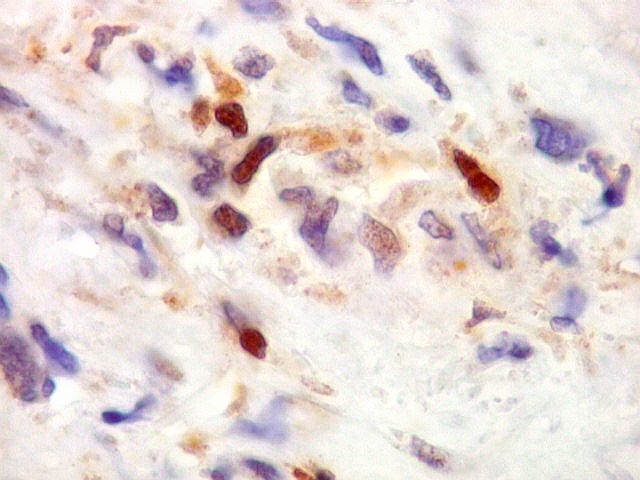
| HHF-35. Positividade citoplasmática em células neoplásicas isoladas, na maioria perivasculares. Negativo na grande parte das células tumorais e nas células gigantes multinucleadas tipo osteoclasto | CD56. Positivo em padrão membrana em parte das células neoplásicas | S-100. Positivo em células isoladas, negativo na maior parte das células neoplásicas. |
 |
 |
 |
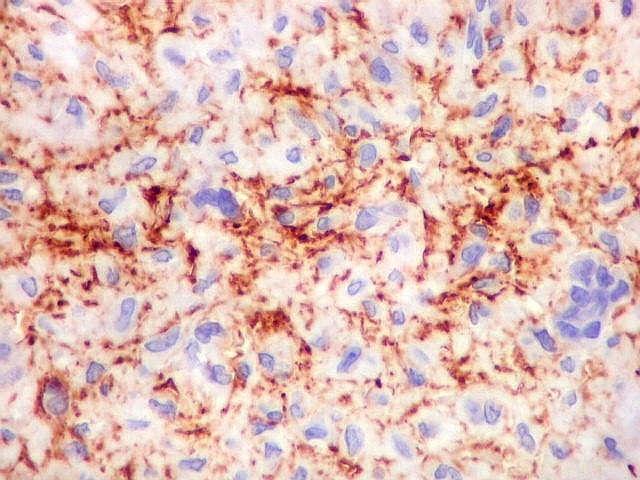
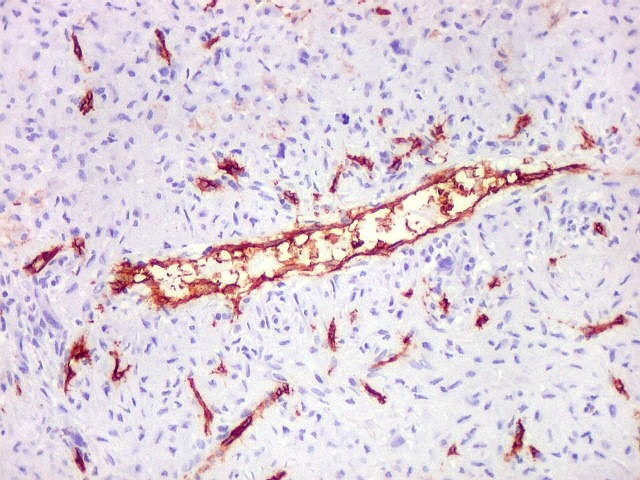
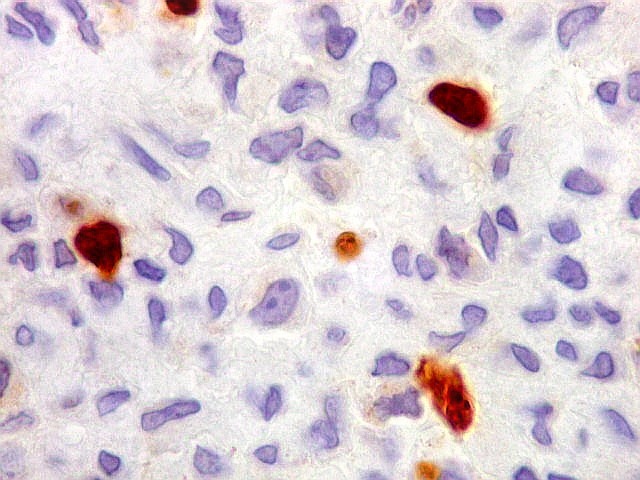
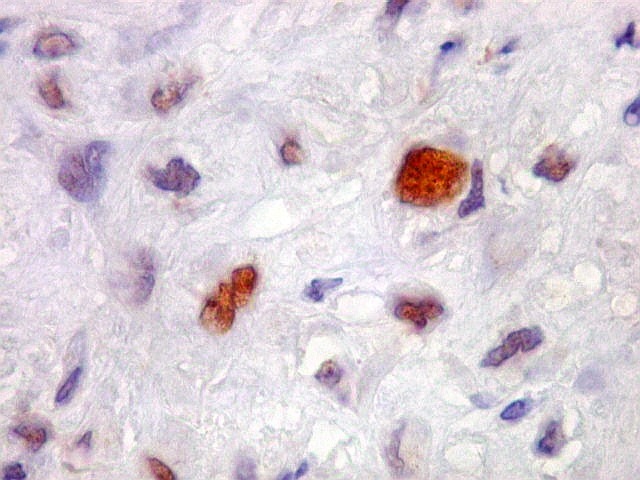
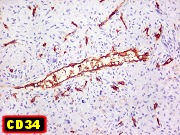
| CD34. Positivo em vasos, negativo no tumor. Demonstra rede capilar extensa e bem distribuída | Ki-67. Positividade em pequena proporção dos núcleos das células neoplásicas. Células tipo osteoclasto sempre negativas. | p53. Positivo em poucas células neoplásicas. Células tipo osteoclasto continham simultaneamente núcleos marcados e negativos. |
 |
 |
 |
| Destaques da microscopia eletrônica. | ||
|
|
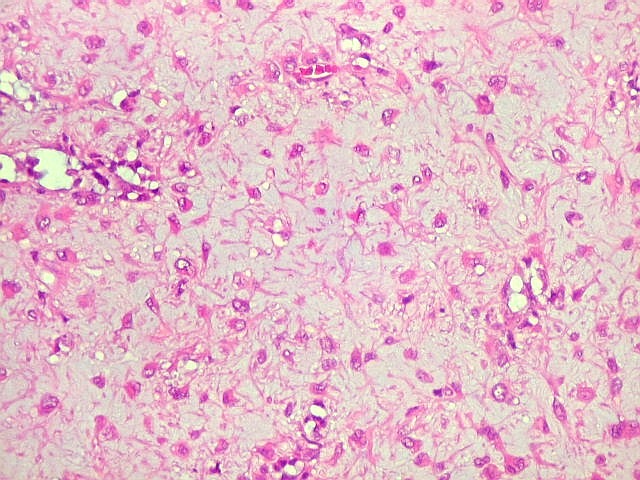
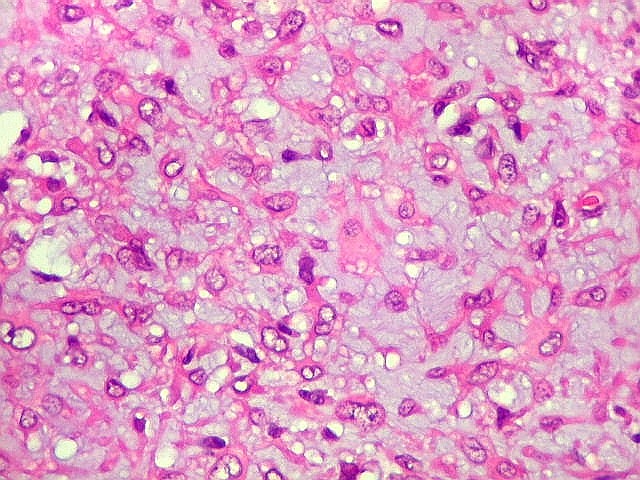
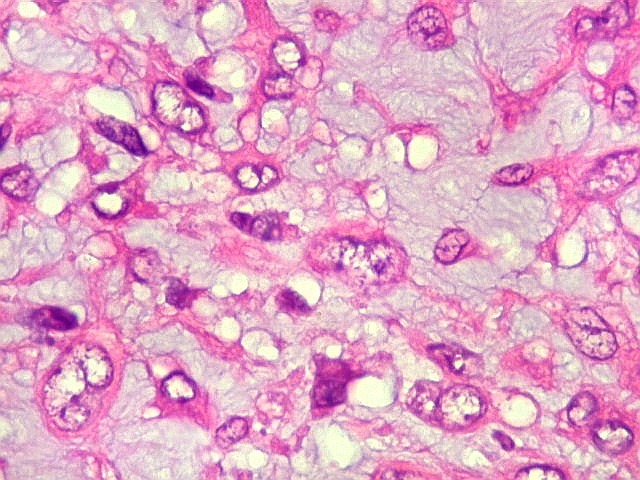
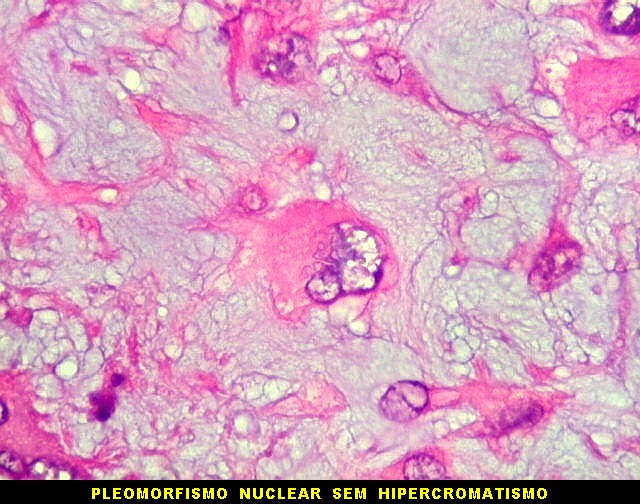
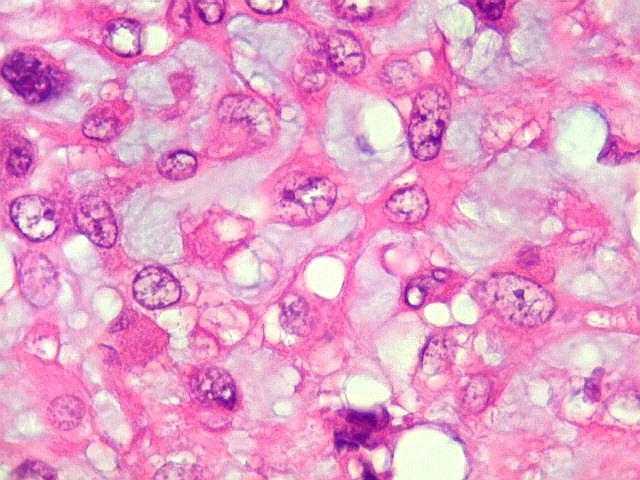
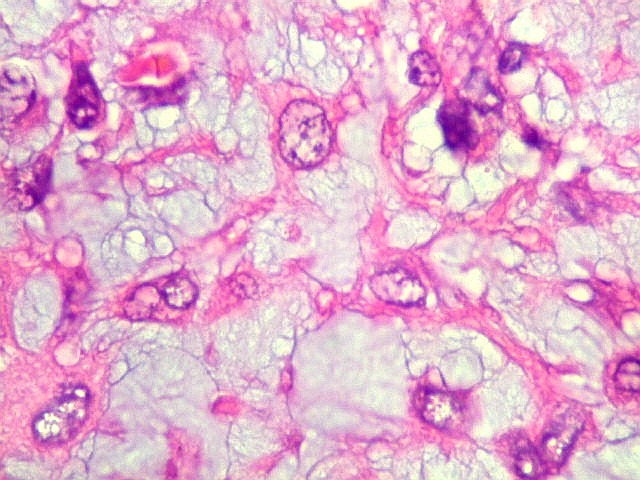
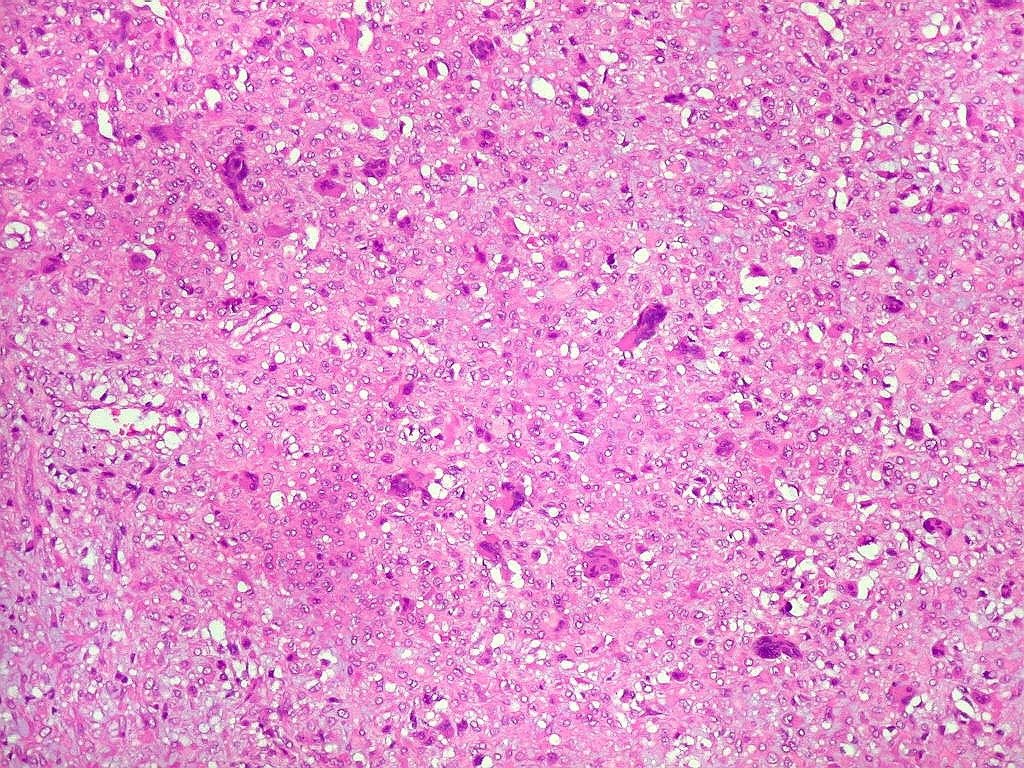
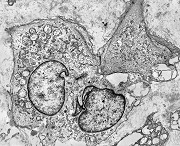
| Aspecto geral do tumor. Neoplasia mesenquimatosa composta por células poligonais ou estreladas em matriz amorfa levemente basófila, permeada por pequenos vasos de paredes finas. Estes são melhor visualizados na imunohistoquímica para CD34. As células são polimórficas, com moderadas atipias nucleares e freqüente binucleação. Contudo, não se notam núcleos hipercromáticos, figuras de mitose ou necrose. Células gigantes multinucleadas do tipo osteoclasto são mais freqüentes em certas áreas. |
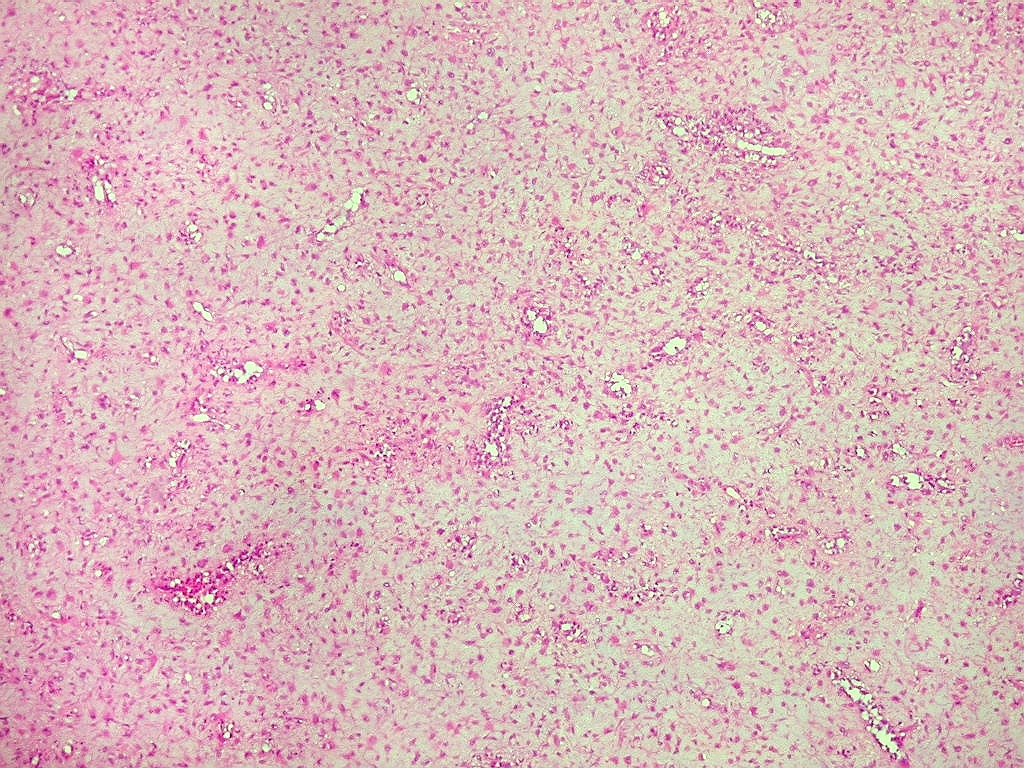 |
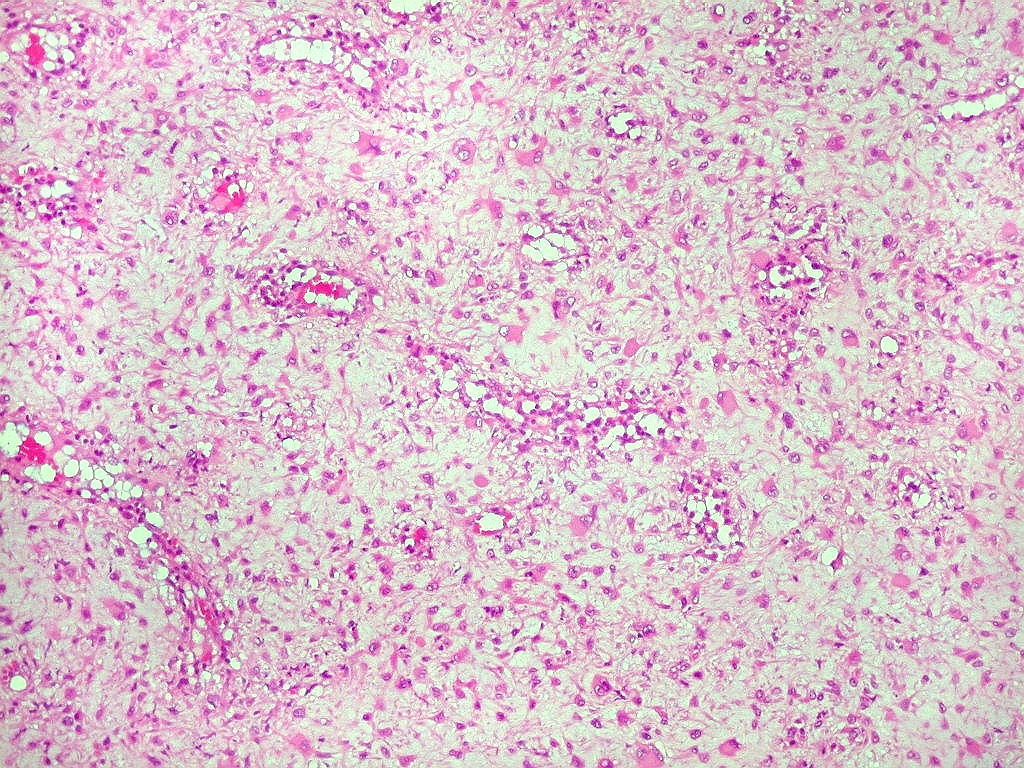 |
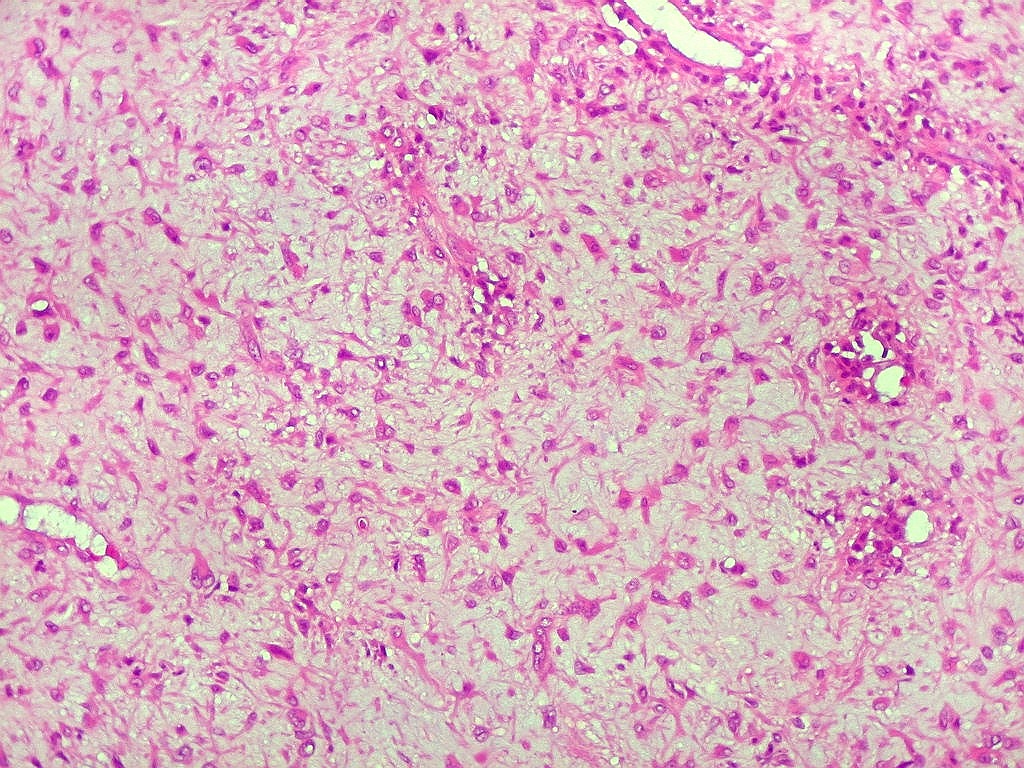 |
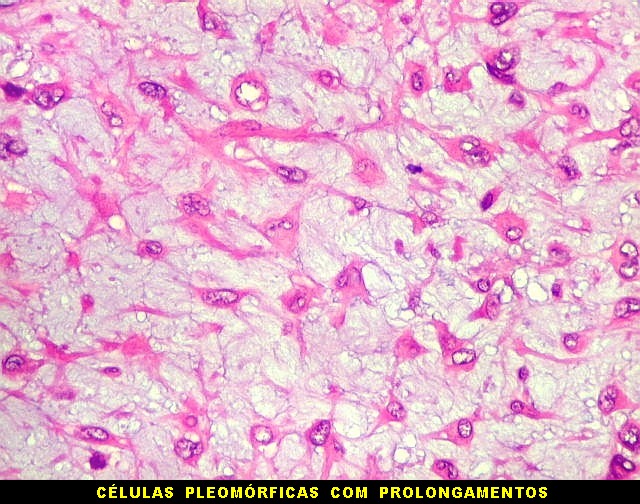
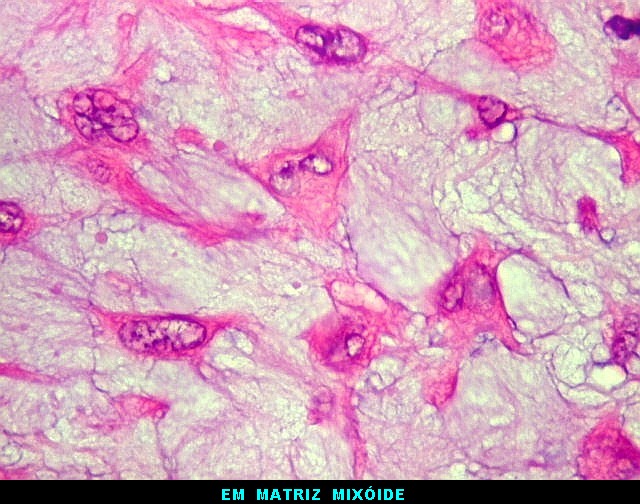
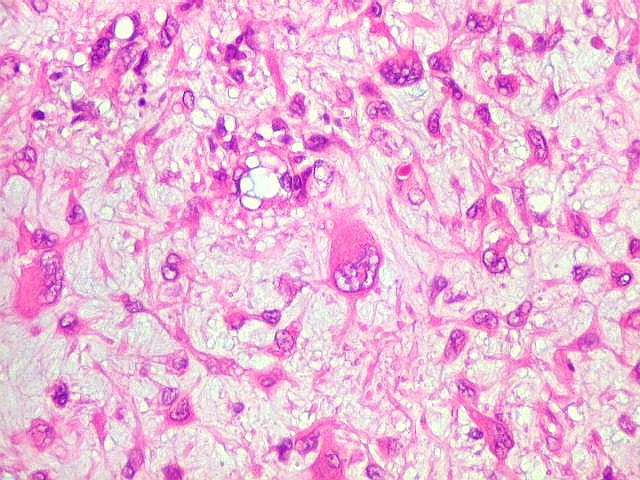
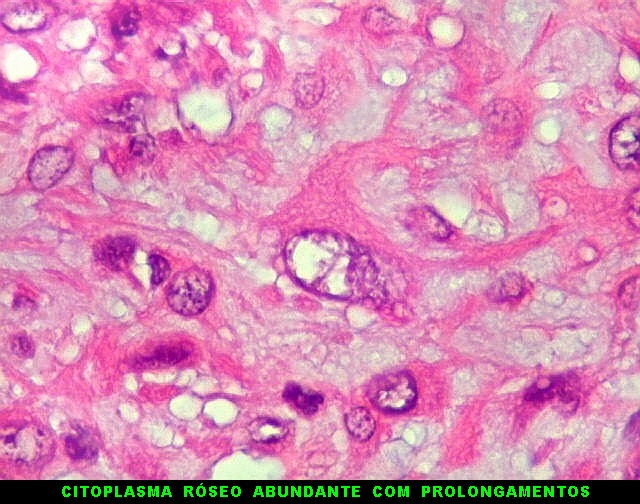
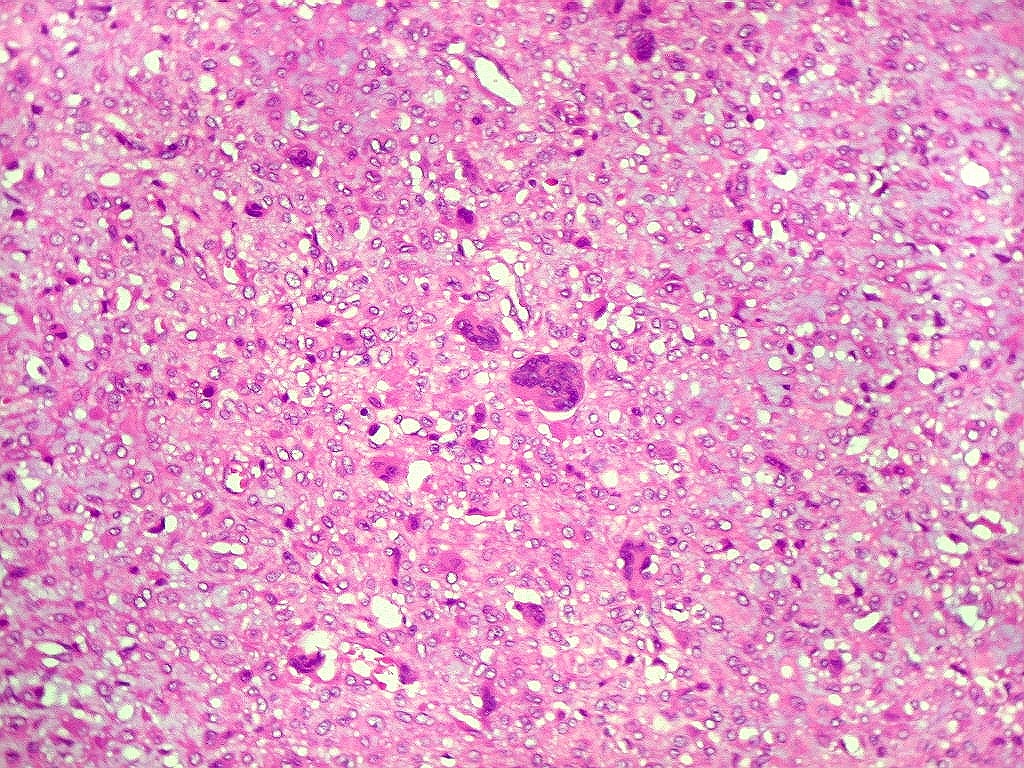
| Aspecto mixóide com polimorfismo celular. No fibroma condromixóide, como o nome indica, descreve-se coexistência de dois padrões de tecido, cartilaginoso e mixóide (ver texto). No presente exemplo, contudo, há predominância praticamente total das áreas mixóides. As células nestas áreas têm contorno irregular com múltiplos prolongamentos citoplasmáticos finos que atravessam a matriz e tendem a tocar os prolongamentos de células vizinhas. Estas relações são melhor observadas em microscopia eletrônica. | |
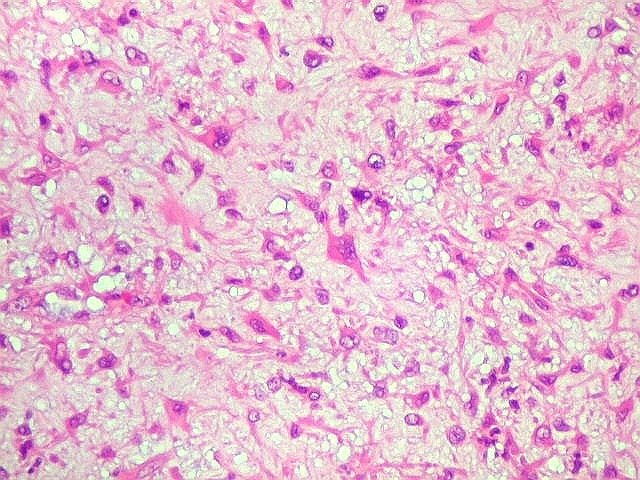 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
|
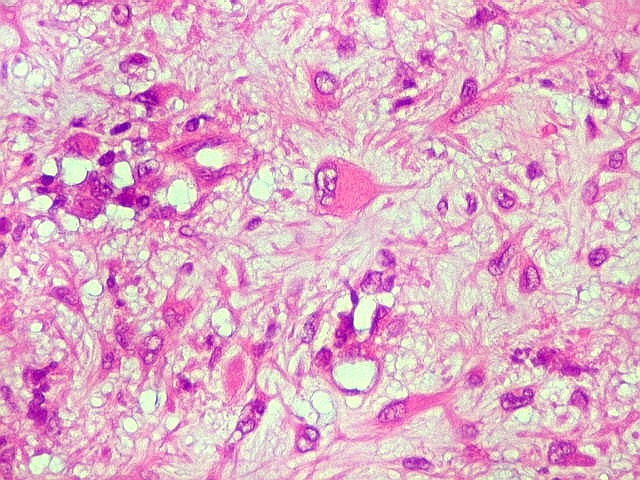
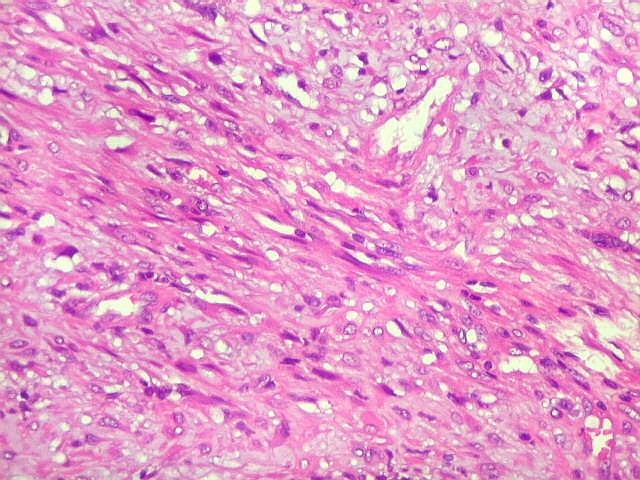
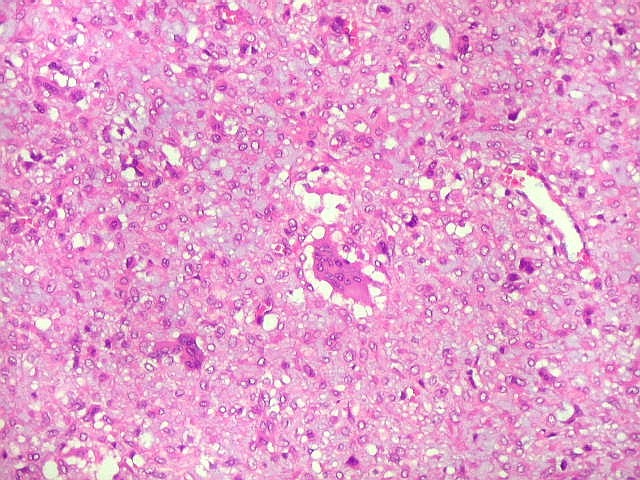
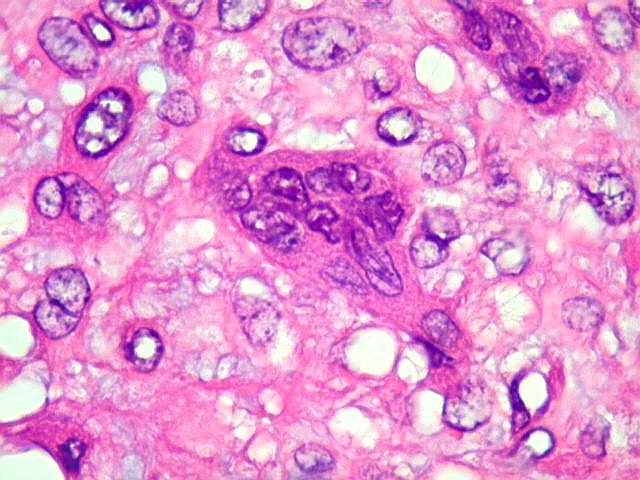
| Área de células fusiformes. Faixas de células alongadas separavam pseudolóbulos tumorais. | |
 |
 |
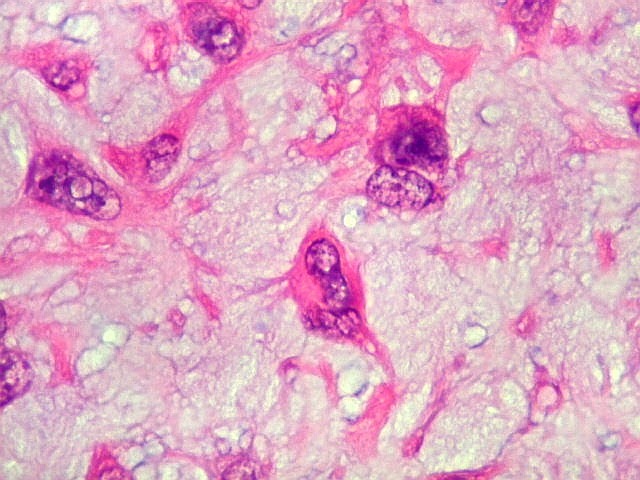
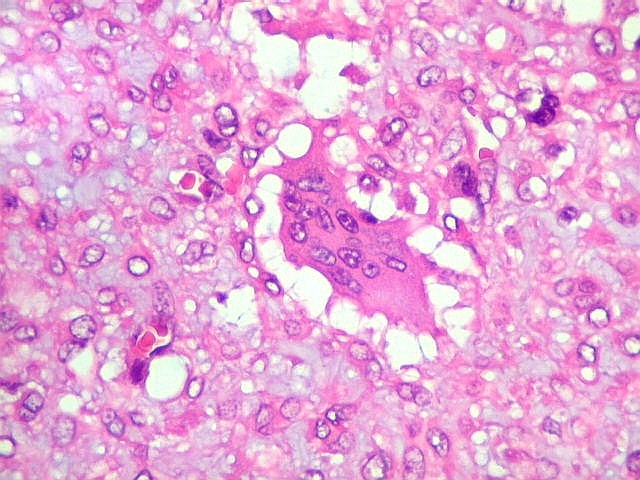
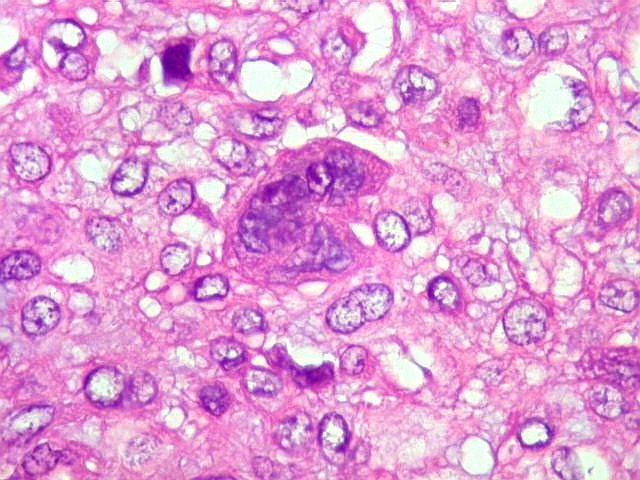
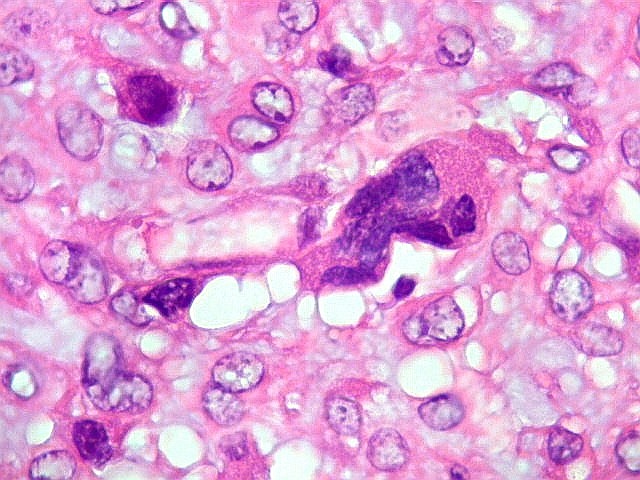
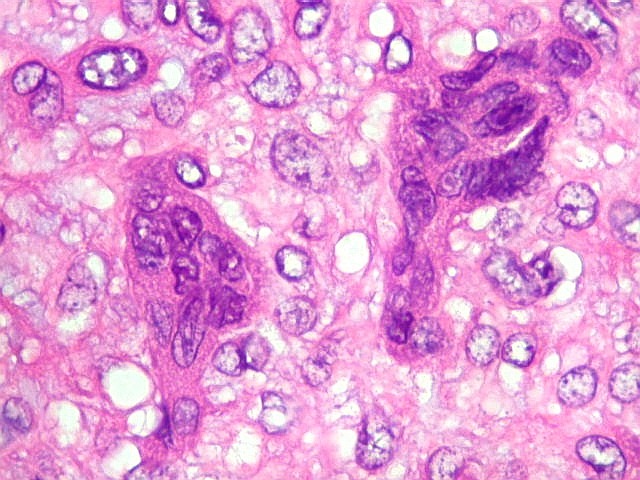
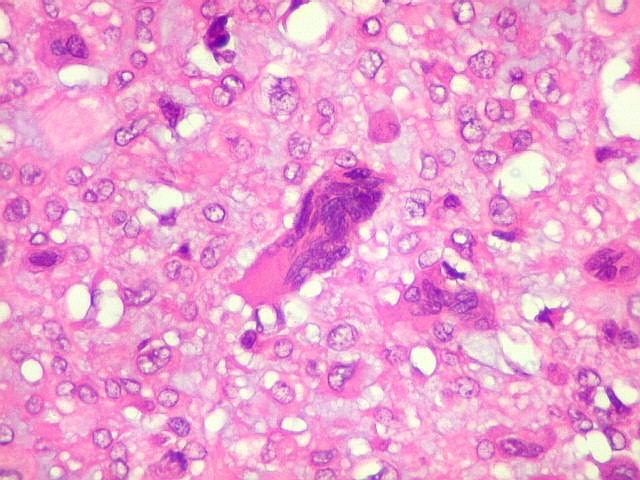
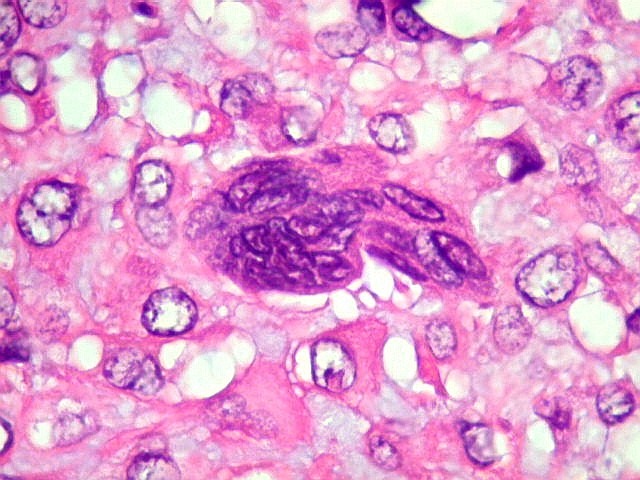
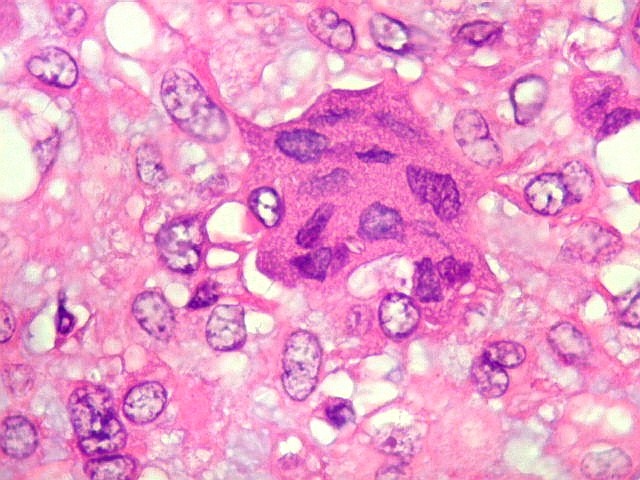
| Células
gigantes multinucleadas do tipo osteoclasto.
São elementos não neoplásicos participantes do tumor. Contêm vários núcleos, ao contrário das células neoplásicas, e o citoplasma é bem delimitado, sem prolongamentos. Ocorrem esparsamente, mas concentram-se entre os pseudolóbulos tumorais. |
 |
 |
 |
 |
 |
 |
 |
|
 |
 |
 |
 |
 |
 |
 |
 |
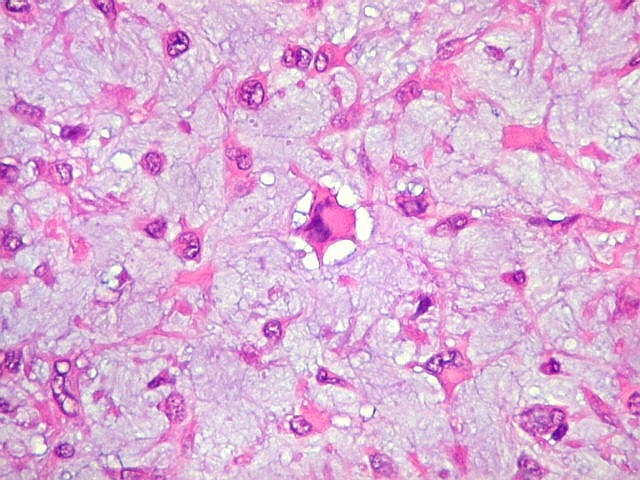
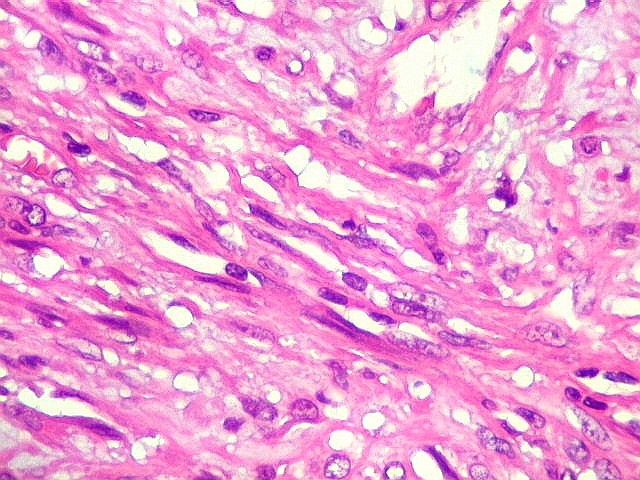
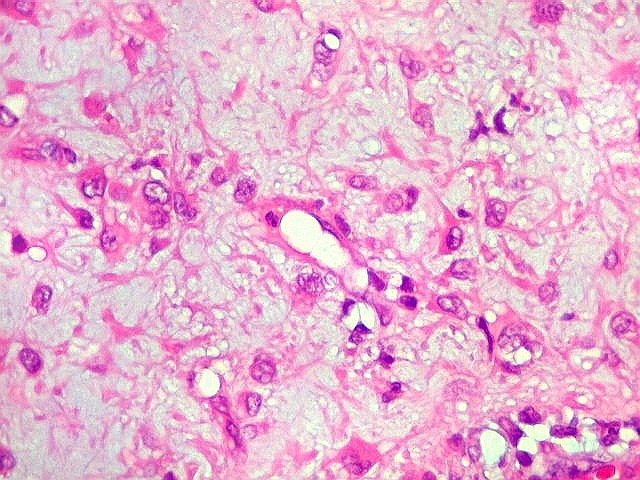
| Vasos. Numerosos, finos, de paredes delgadas. | |
 |
 |
 |
|
| Fibroma
condromixóide.
Definição. Fibroma condromixóide é o mais raro dos tumores que formam cartilagem e, devido à sua morfologia altamente pleomórfica, pode ser confundido com sarcoma. É um tumor benigno, composto de tecido mesenquimatoso mixóide imaturo com diferenciação cartilaginosa incipiente. Tem predileção pelas regiões metafisárias de ossos longos tubulares dos membros inferiores, freqüentemente em situação excêntrica. O termo e conceito atual do tumor foram propostos por Jaffe e Lichtenstein em 1948. Incidência. É raro, correspondendo a < 1% dos tumores ósseos, muito menos comum que o condroblastoma. Tem predileção pelo sexo masculino (1,5 : 1) e segunda e terceira décadas (pico de incidência entre 10 e 30 anos). Há casos até os 50 anos. Localização. Cerca de 1/3 dos casos são observados ao redor do joelho, com maior incidência na metáfise proximal da tíbia, seguida pela metáfise distal do fêmur. Pode também ocorrer nos ossos pequenos dos pés. Membros superiores são raramente acometidos. A pelve, especialmente o ilíaco, é o osso chato mais afetado. Há relatos isolados em ossos craniofaciais, costelas, coluna e ossos tubulares curtos das mãos e pés. Na realidade, pode ocorrer em qualquer osso. Clínica. Dor é o sintoma mais comum e pode durar vários anos. Pode haver aumento de volume dos tecidos moles sobre a lesão. Radiologia. São lesões de aspecto benigno, metafisárias, excêntricas, ovais, com maior eixo paralelo ao do osso. Podem estender-se às regiões limítrofes (epifisárias e diafisárias), mas é incomum que o tumor atravesse uma placa de crescimento aberta. As margens intramedulares do tumor são caracteristicamente nítidas, irregulares e escleróticas. As lesões são geralmente totalmente líticas, raramente com calcificações na matriz. É mais fácil observar calcificações na histologia que na radiografia simples. Pode haver expansão excêntrica do córtex e, quando marcada, pode representar formação de um cisto ósseo aneurismático secundário. Nos ossos pequenos das mãos e pés, os fibromas condromixóides são geralmente centrais, expandindo o osso uniformemente. Nos ossos chatos, p. ex. pelve, as lesões podem ser grandes, de contorno irregular e com loculações intralesionais proeminentes. Macro. Massa lobulada branco-acinzentada, firme, bem delimitada, sempre confinada pelo periósteo intacto. Pode haver áreas semisólidas ou mixóides, mas os focos condróides geralmente não são observados macroscopicamente. Micro. Tipicamente tem arquitetura pseudolobulada, com áreas mixomatosas e condróides separadas por zonas de tecido mononuclear hipercelular contendo células gigantes multinucleadas esparsas. O componente básico da lesão é o tecido mesenquimatoso mixóide imaturo que sofre diferenciação cartilaginosa. As células tumorais dentro dos pseudolóbulos podem ser pleomórficas e mostrar hipercromatismo e multinucleação, levantando o diagnóstico diferencial com condrossarcoma. Contudo, mitoses são extremamente raras no fibroma condromixóide. A celularidade das áreas mixóides, compostas por células estreladas ou fusiformes, aumenta na periferia das mesmas. Nestas áreas, as células são estreladas, e seus delicados processos citoplasmáticos estendem-se através da substância fundamental mucinosa, aproximando-se ou fazendo contacto com as células vizinhas. As áreas mais solidamente celulares são compostas de células mononucleares representando condroblastos. Nestas áreas cartilaginosas, as células neoplásicas estão situadas em lacunas. Células gigantes multinucleadas esparsas do tipo osteoclasto, não neoplásicas, podem intervir entre os pseudolóbulos. Áreas de diferenciação cartilaginosa mais madura, com calcificações finas ou grosseiras, podem ocorrer. Imunohistoquímica. Há positividade variável para S-100, mais forte nas áreas com diferenciação condroblástica mais madura e mais fraca ou focal nas áreas mixóides frouxas. Estes achados indicam que o fibroma condromixóide é de natureza cartilaginosa, mas representa um estágio mais precoce e menos maduro de diferenciação cartilaginosa que o condroblastoma. Tratamento e prognóstico. O tratamento de escolha é ressecção em bloco do tumor. Curetagem provavelmente deixará algum tumor residual, que pode recidivar. A taxa de recidiva é de cerca de 20% e não se correlaciona com nenhum dado ou feição histológica. Radioterapia tem sido usada para tratar tumores cirurgicamente inacessíveis. Uma complicação descrita é aparecimento de um tumor maligno, como sarcomas ou fibrohistiocitoma maligno. Contudo, malignização espontânea não parece ocorrer no fibroma condromixóide. Fontes.
|
| Agradecimentos. Caso gentilmente contribuído pelos Drs. Antonio Augusto Roth Vargas, Marcelo Senna Xavier de Lima, Paulo Roland Kaleff e Marcel Ramos Olivatto, Hospital Santa Casa de Limeira, Limeira, SP. Estudado conjuntamente com a Profa. Dra. Eliane Maria Ingrid Amstalden, especialista em Patologia Óssea e de Partes Moles do Depto de Anatomia Patológica da FCM-UNICAMP. Preparações histológicas pelos técnicos Aparecido Paulo de Moraes e Viviane Ubiali. |
| Para mais imagens deste caso, e textos : fibroma condromixóide, fibronectina, integrinas | TC / RM | HE | |
 |
 |
||
| Colorações especiais | IH - VIM, 1A4, HHF-35 | IH - CD56, S100, CD34, Ki67, p53 | ME |
 |
 |
 |
 |
|
imagem, patologia |
|
|
| Neuropatologia
- Graduação |
Neuropatologia -
Estudos de casos |
Neuroimagem
- Graduação |
Neuroimagem -
Estudos de Casos |
Roteiro
de aulas |
Textos
de apoio |
Correlação
Neuropatologia - Neuroimagem |
| Índice alfabético - Neuro | Adições recentes | Banco de imagens - Neuro | Textos ilustrados | Neuromuscular | Patologia - outros aparelhos | Pages in English |
|
|
|
|