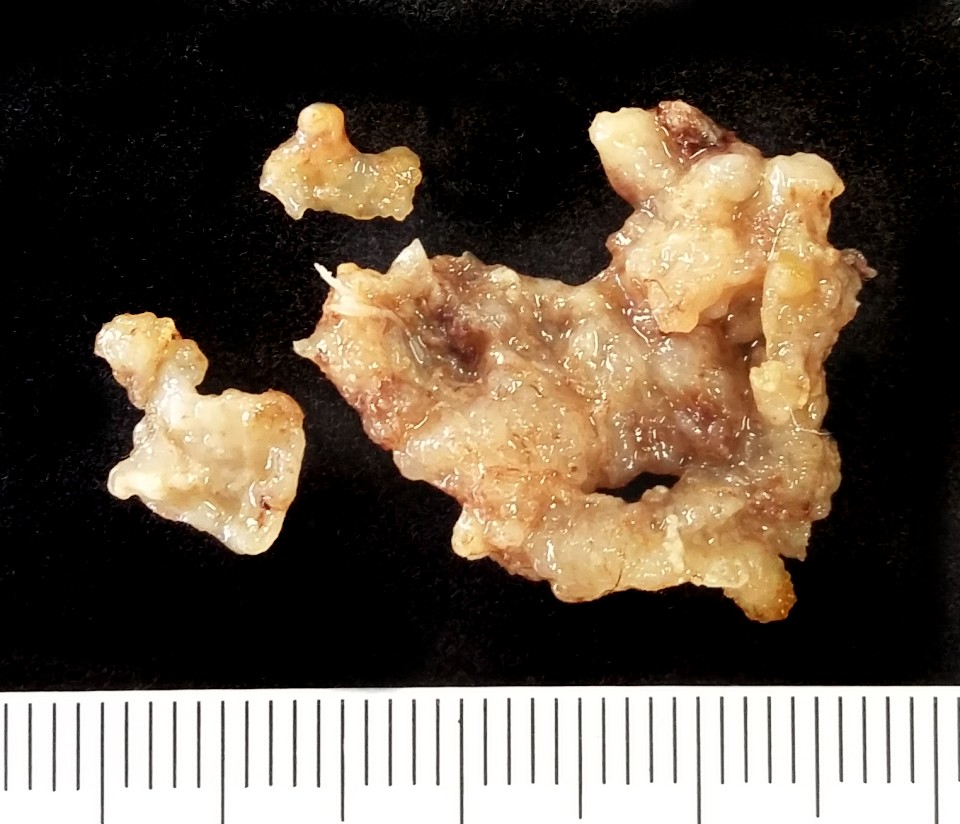
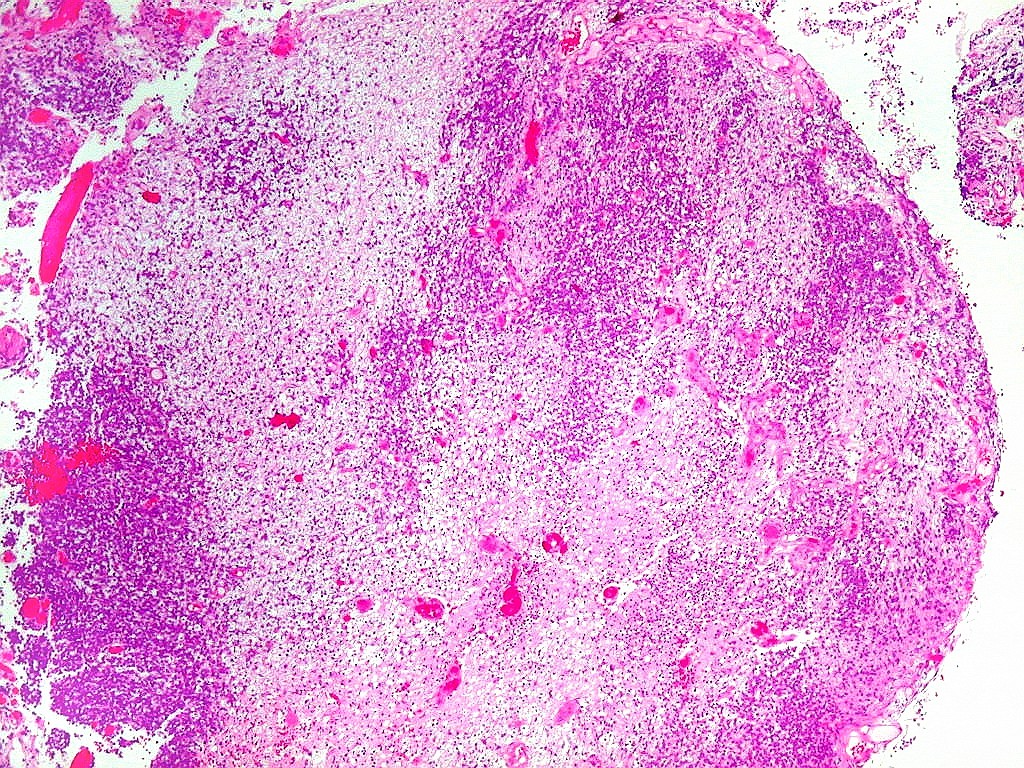
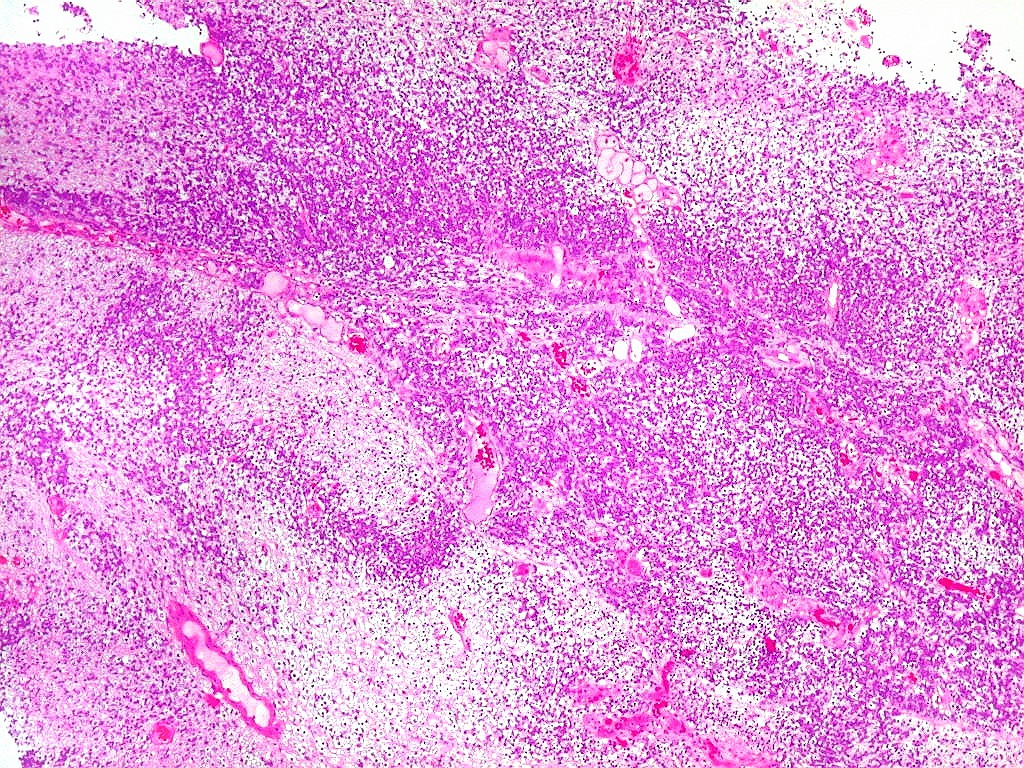
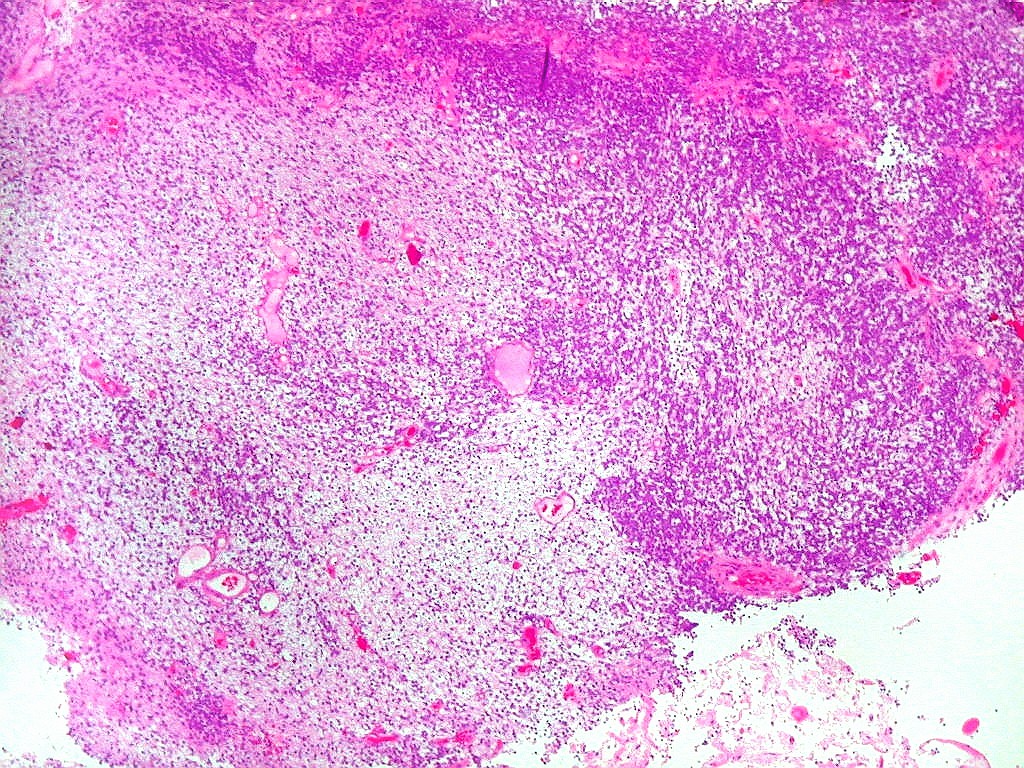
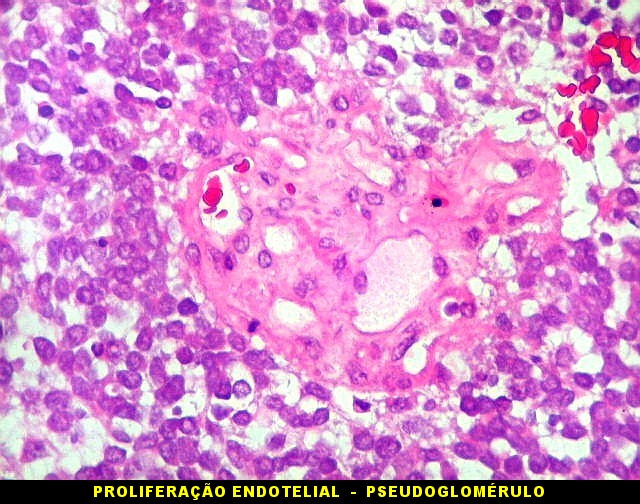
| Tumor teratóide
/ rabdóide atípico.
Genética.
Gene SMARCB1 = INI1 (integrase inactivator -1), BAF47 (BRM ou BRG1 associated
factors subunit of 47 kDa). Subunidade central de um complexo remodelador
de cromatina dependente de ATPase composto de várias subunidades.
Tem feições de um gene supressor tumoral, com papel chave
na ativação ou repressão de transcrição
gênica através de remodelação de cromatina.
Está localizado no cromossomo 22q11.23 e é expressado universalmente
em todas células nucleadas de mamíferos. Perda da expressão
reflete inativação bialélica através de mutações
ou deleções que podem ocorrer sem ou com mutação
de linhagem germinativa predisponente.
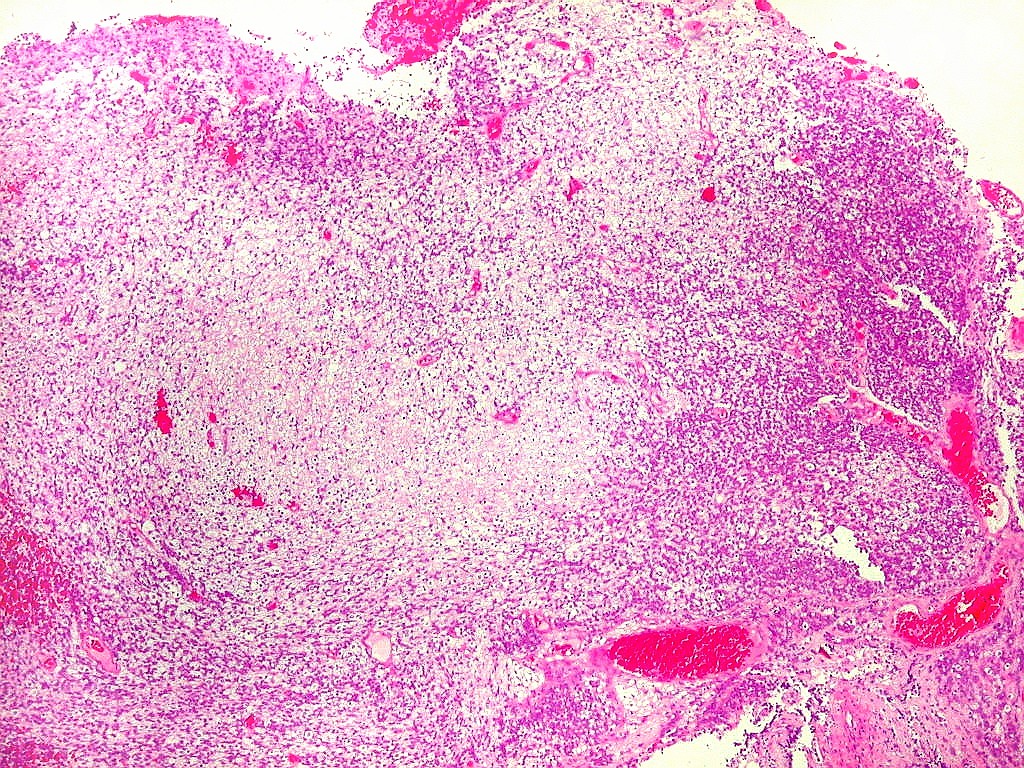
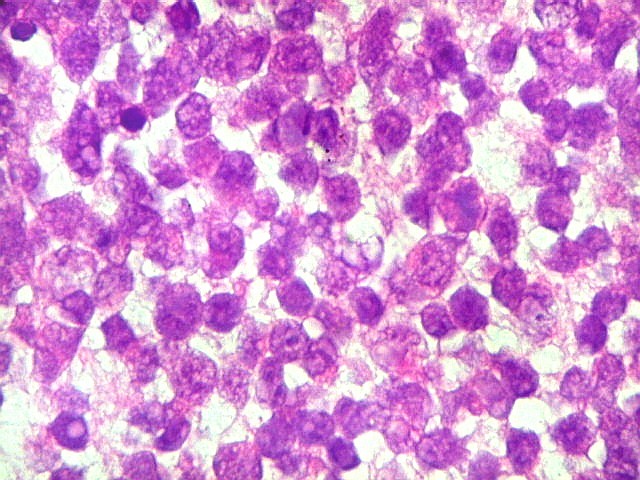
Diagnóstico histológico.
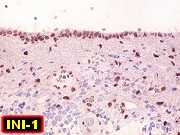
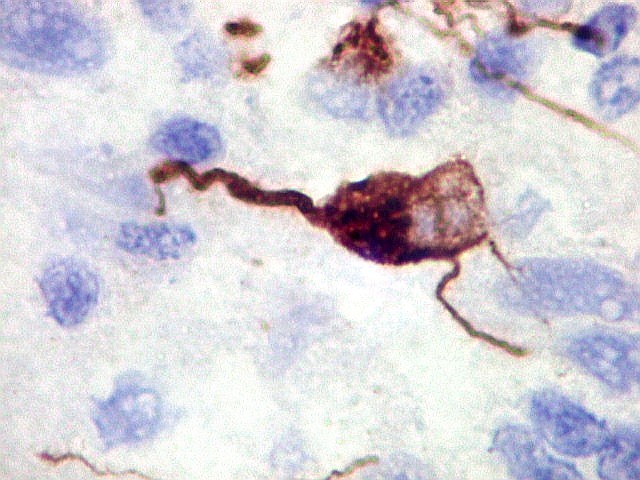
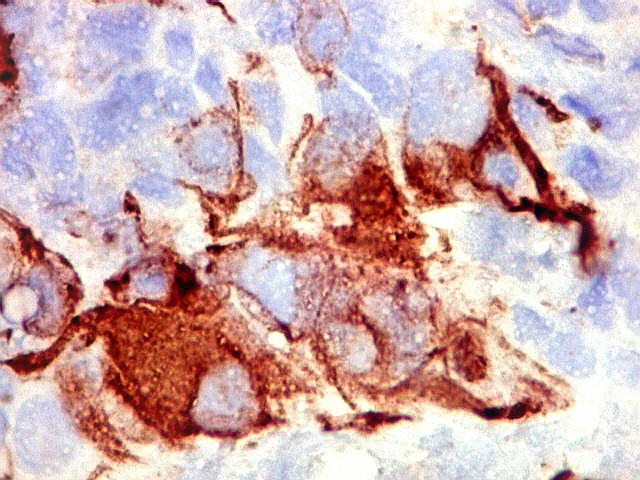
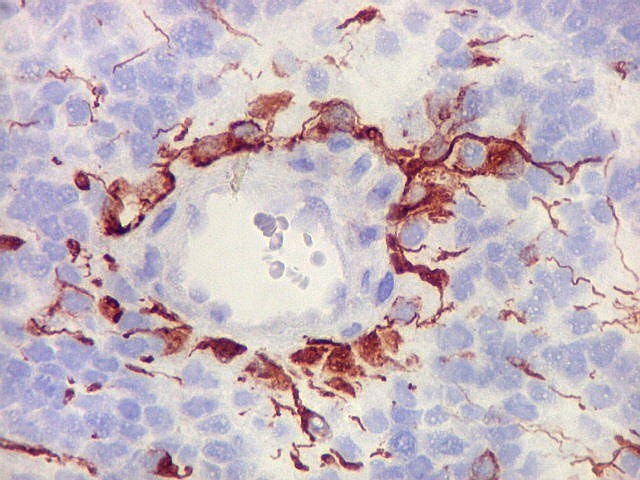
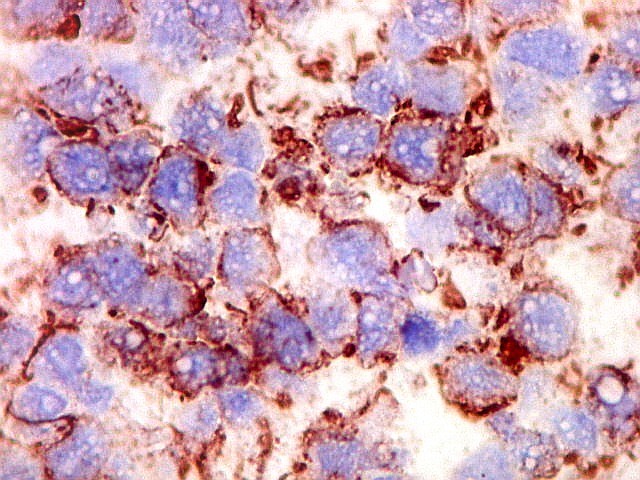
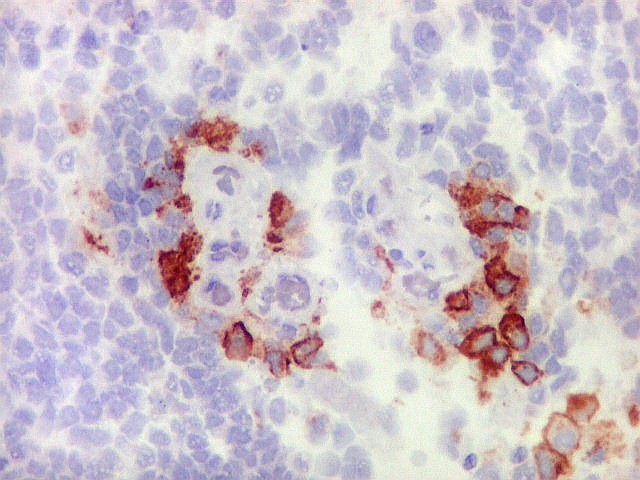
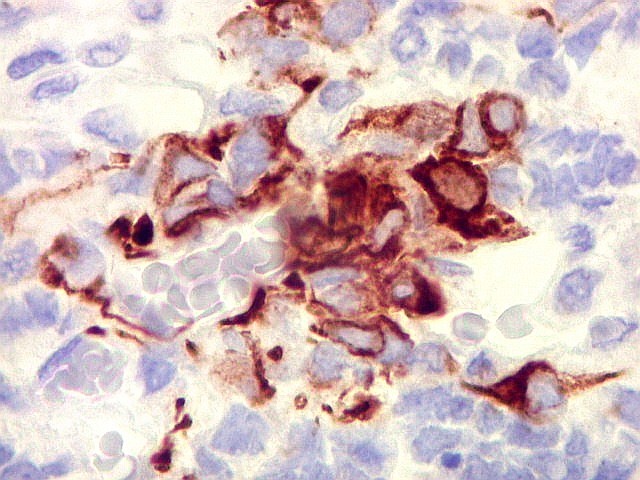
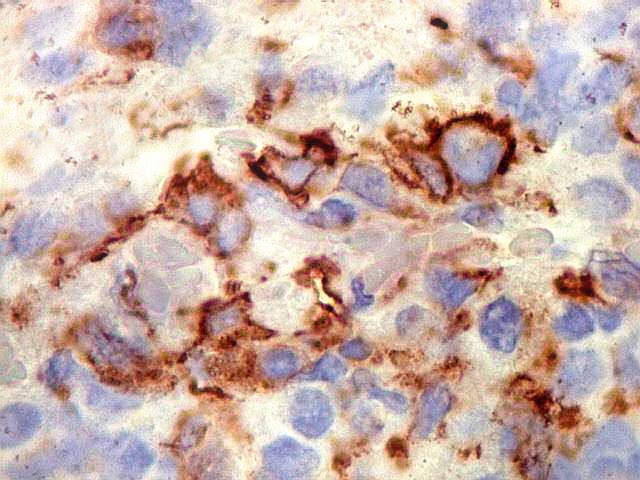
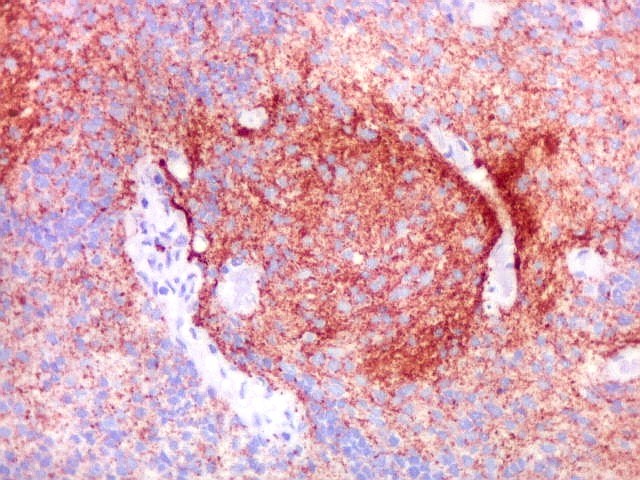
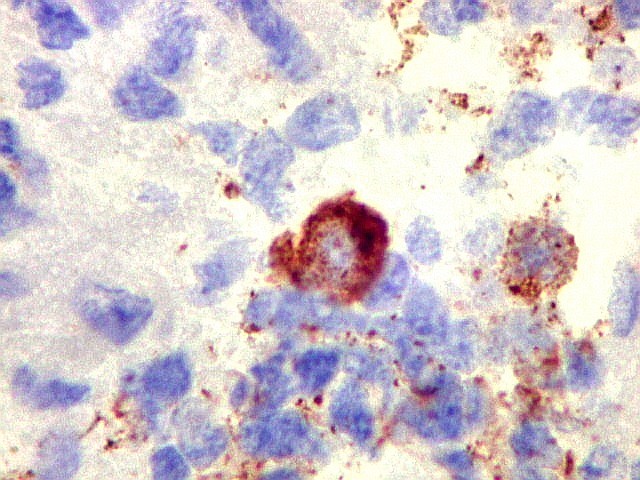
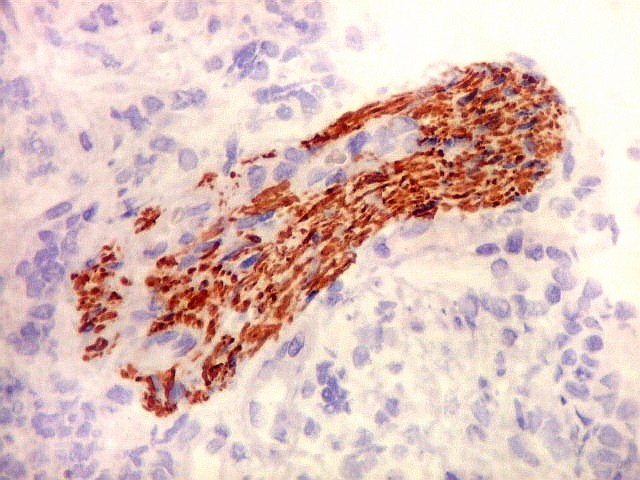
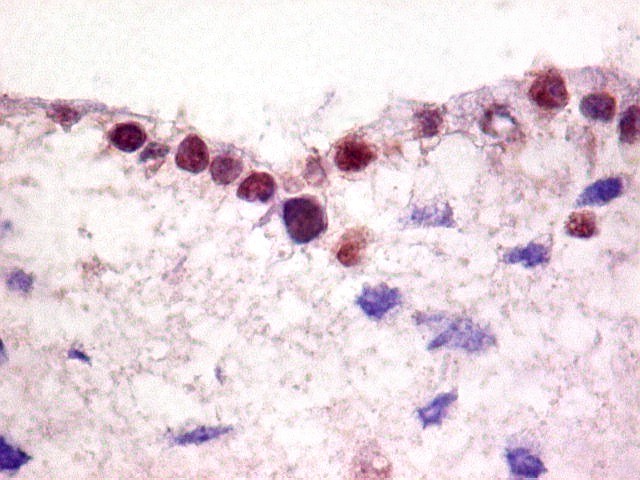
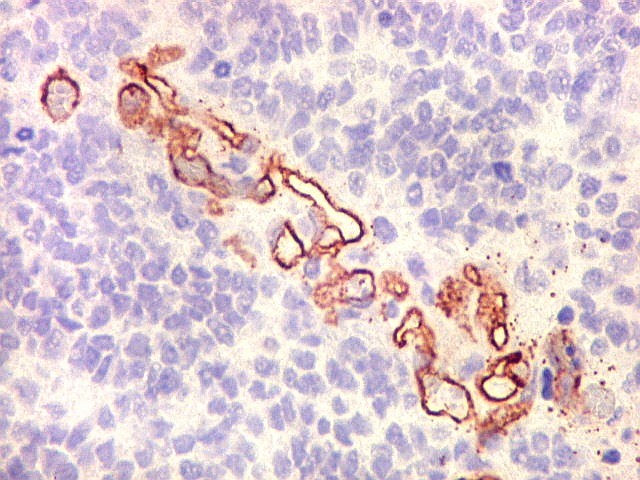
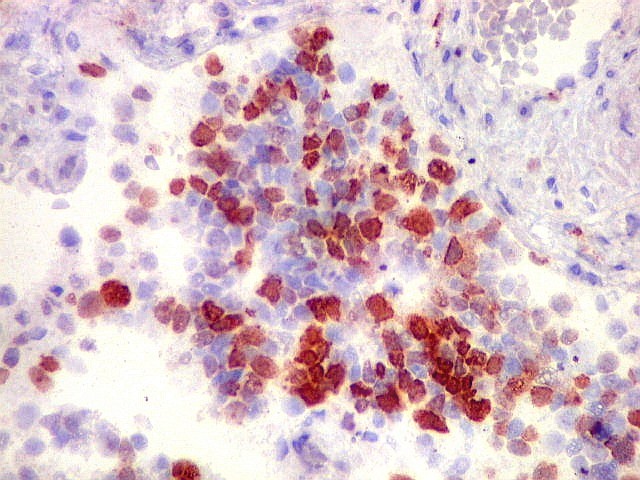
Demonstração por imunohistoquímica da ausência
de reação para INI-1 é fundamental para o diagnóstico.
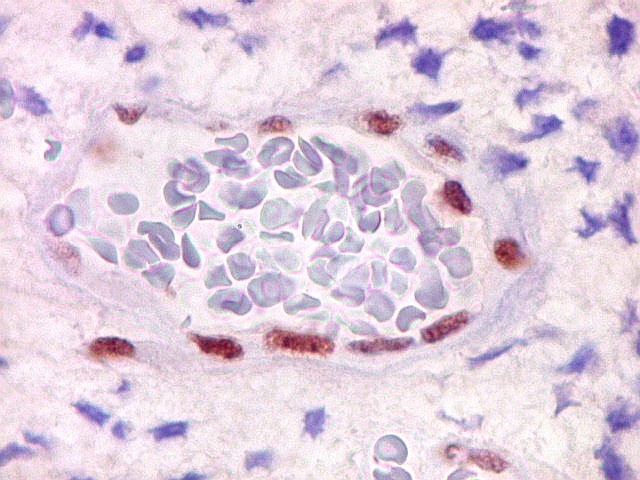
Controle interno está sempre presente na forma de células
do sangue, inflamatórias ou endoteliais. A maioria dos tumores
rabdóides, seja do rim ou o ATRT, mostram imunoperfil polifenotípico
com expressão de marcadores neurais, epiteliais e mesenquimais.
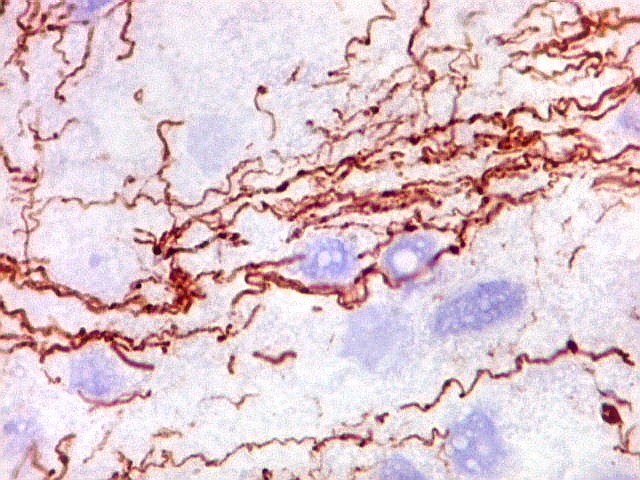
Quase todos expressam vimentina, a maioria expressa EMA e citoqueratinas
de amplo espectro de pesos moleculares. Há expressão variável
de NSE, SNF, GFAP e actina de músculo liso (1A4). Desmina
e CD34 só raramente são expressadas. Cerca de metade dão
positividade padrão membrana para CD99, levantando a dificuldade
diagnóstica com sarcoma de Ewing.
Outros tumores com
perda de INI-1 além do ATRT.
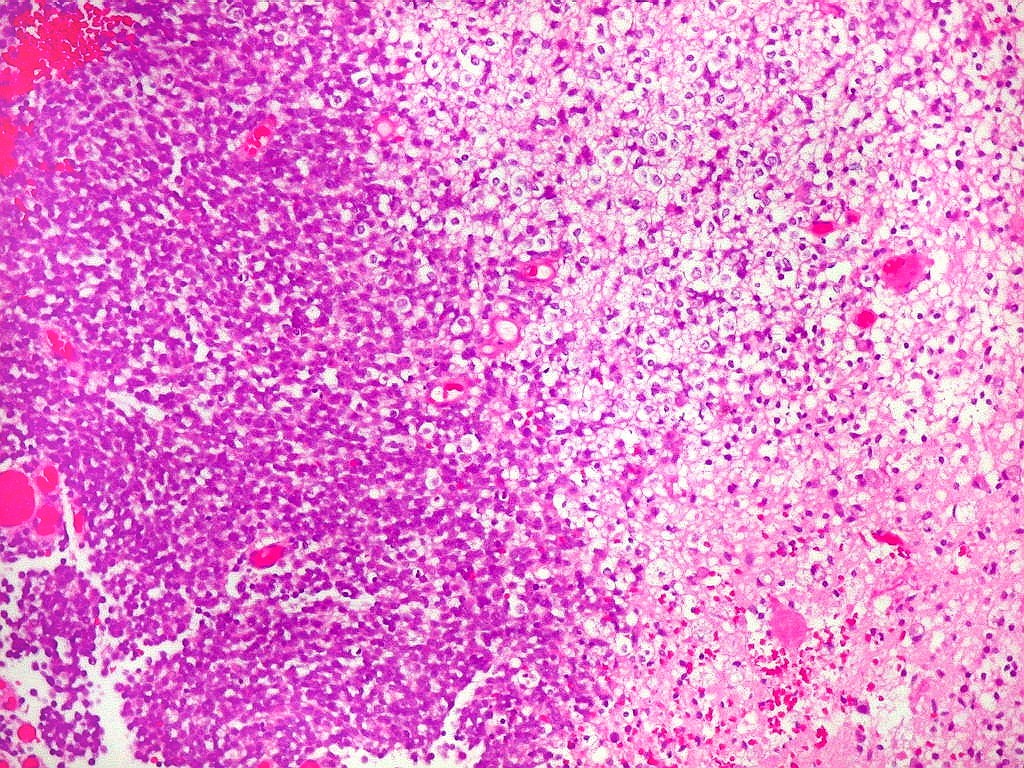
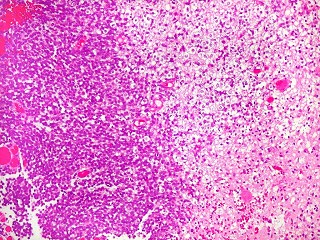
São tumores de crianças muito jovens, e na maioria seguem
curso rápido ao êxito letal. Morfologicamente, a maioria,
mas não todos, contêm uma população de células
rabdóides.
-
Tumor prototípico
tumor rabdóide maligno, primeiro descrito
no rim, mas que ocorre também em tecidos moles e vísceras.
-
Outros tumor
neuroepitelial cribriforme, carcinoma medular renal, sarcoma epitelióide.
-
Tumores com
perda variável de expressão de INI-1 subgrupos do tumor
maligno de baínha de nervos periféricos (MPNST) epitelióide,
schwannomas originados em schwannomatose, subgrupos de cordomas,
carcinomas mioepiteliais, carcinomas sinonasais, sarcoma sinovial.
Ocorrência familial
síndrome da predisposição a tumores rabdóides
(RTPS). RTPS tipo 1 (>98% dos casos) envolve mutações
constitucionais do gene SMARCB1. RTPS tipo 2 (<2% dos casos) envolve
mutações constitucionais do gene SMARCA4. Desenvolvimento
de tumores ocorreria após um second hit envolvendo o outro
alelo. Pacientes com tumores do espectro devem ser avaliados para
mutações do gene na linhagem germinativa, assim como
nos pais, para eventual aconselhamento genético.
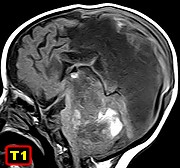
Tumores rabdóides
do SNC.
-
São
por convenção chamados de tumor teratóide/rabdóide
atípico, refletindo a percepção inicial que esses
tumores podem ter áreas de diferenciação mesenquimal
e focos teratóides epiteliais.
-
São
raros, <2% dos tumores cerebrais pediátricos (idade mediana 1,4
anos), ocorrem em proporções semelhantes nos andares supra
e infratentorial e 7% na medula espinal.
-
Prognóstico
muito desfavorável, sobrevida mediana 9 meses após o diagnóstico.
-
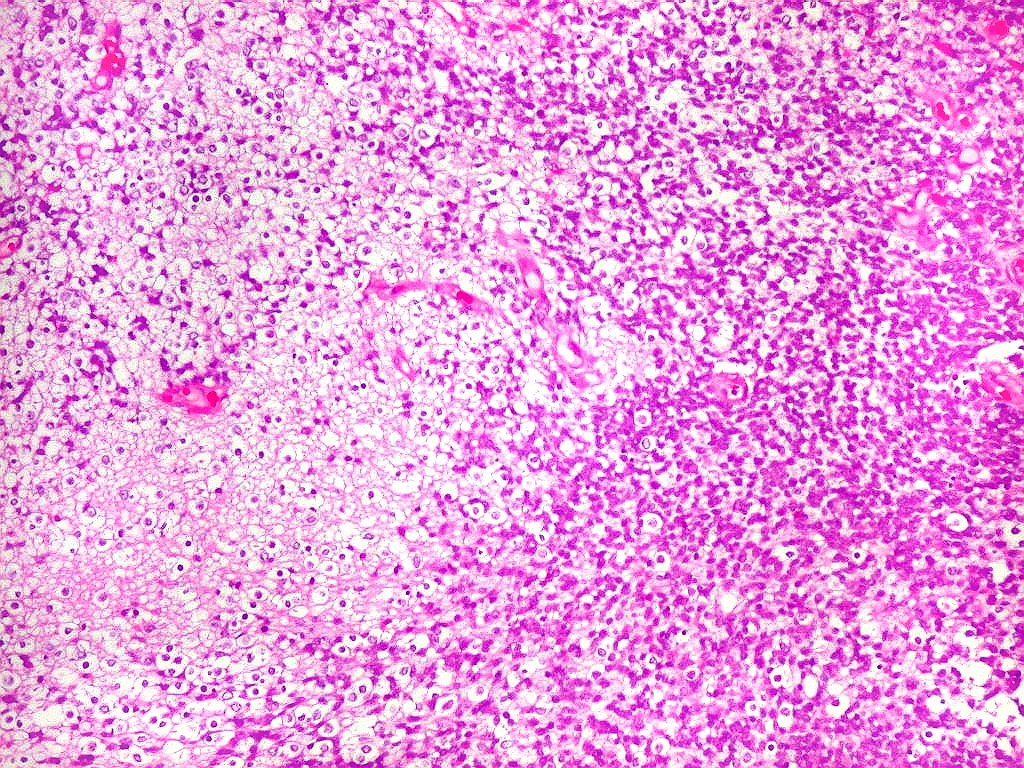
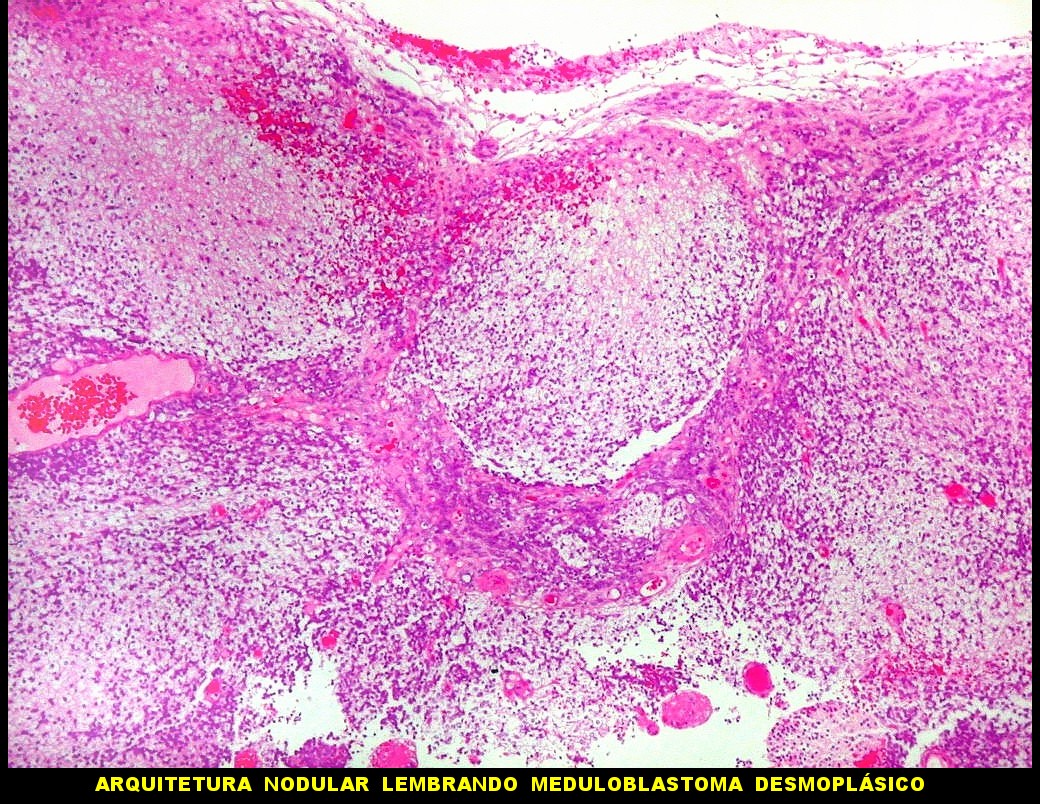
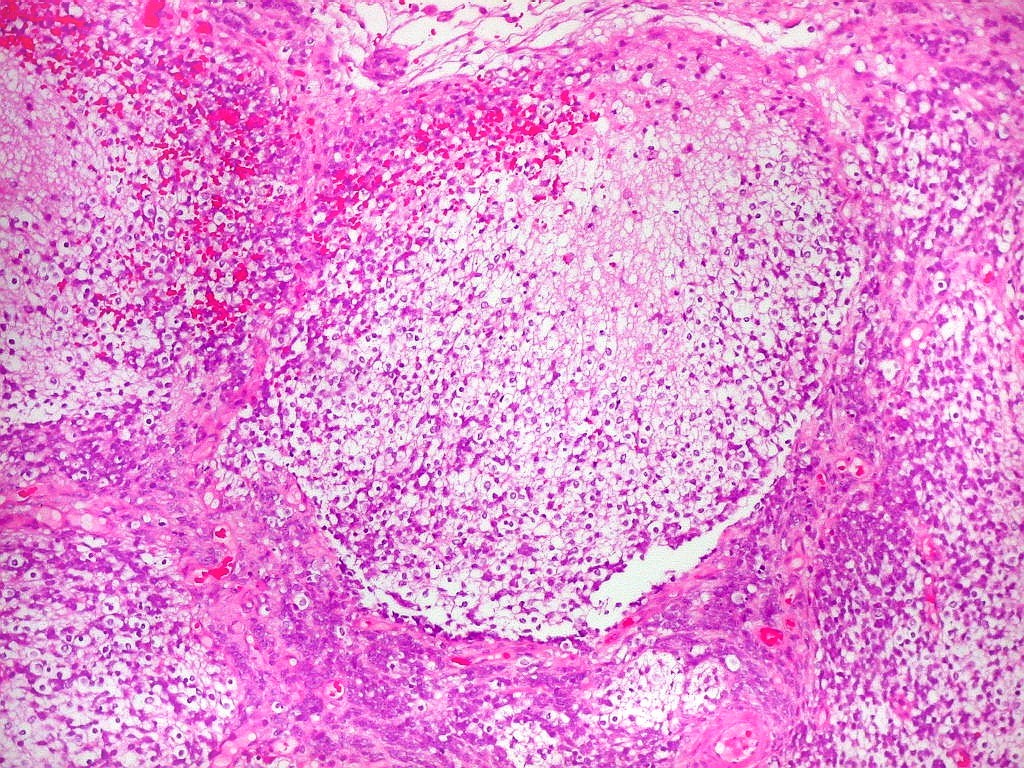
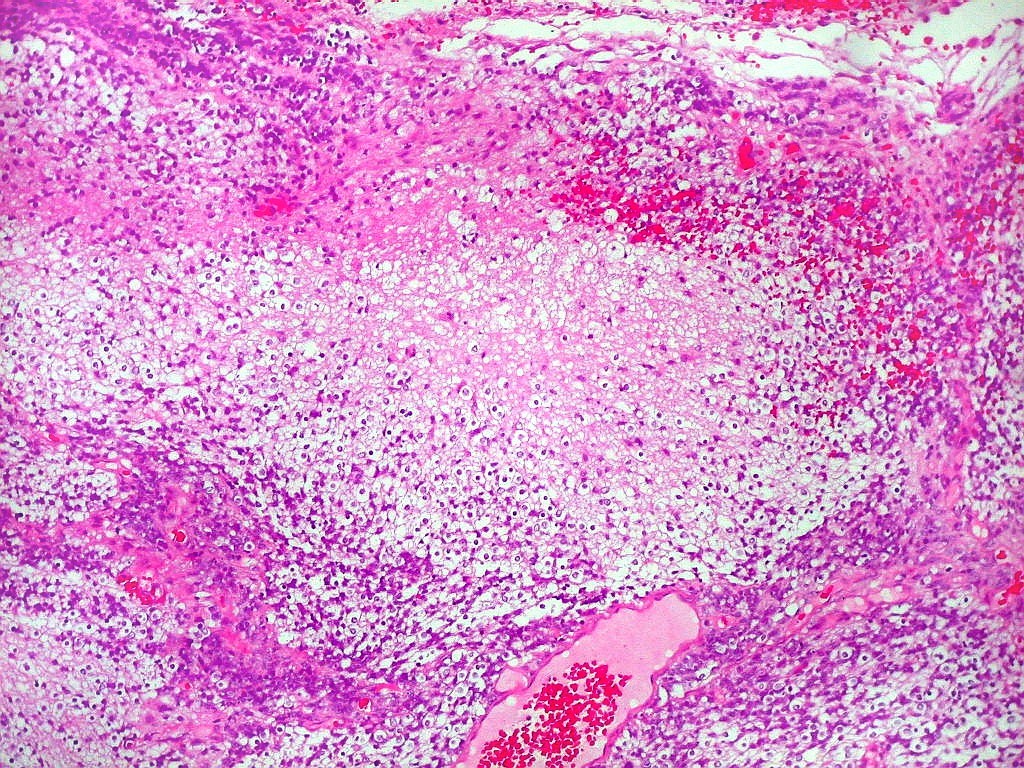
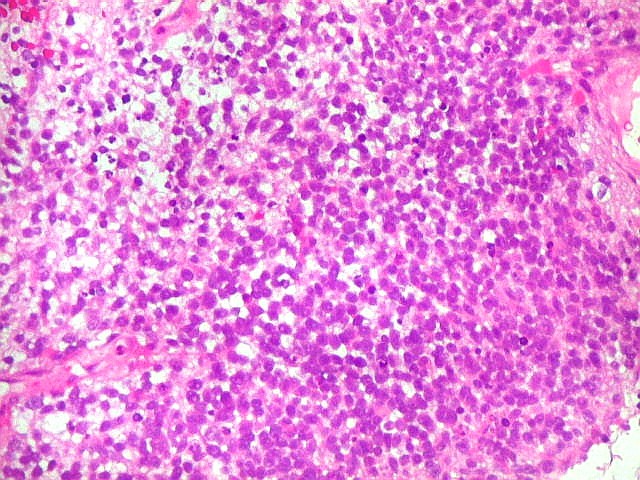
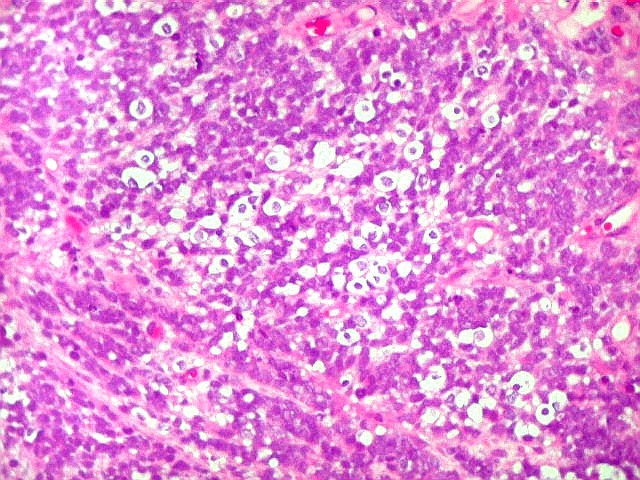
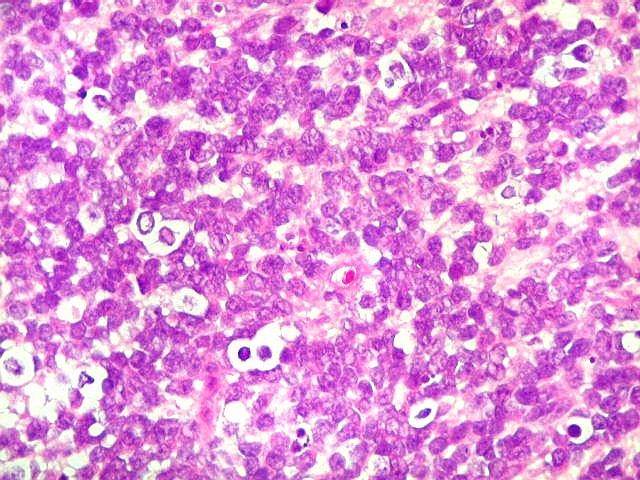
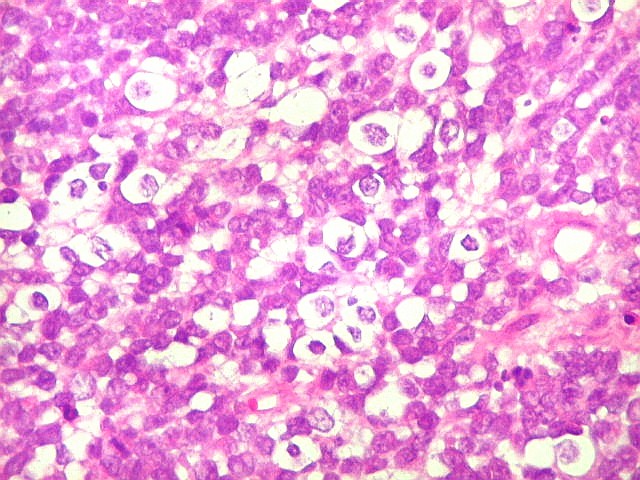
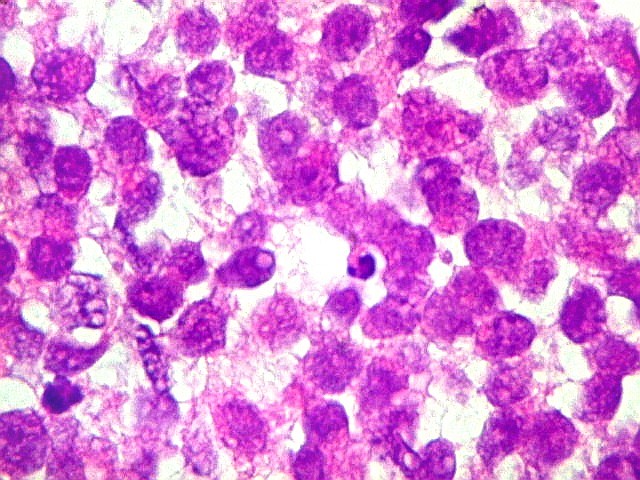
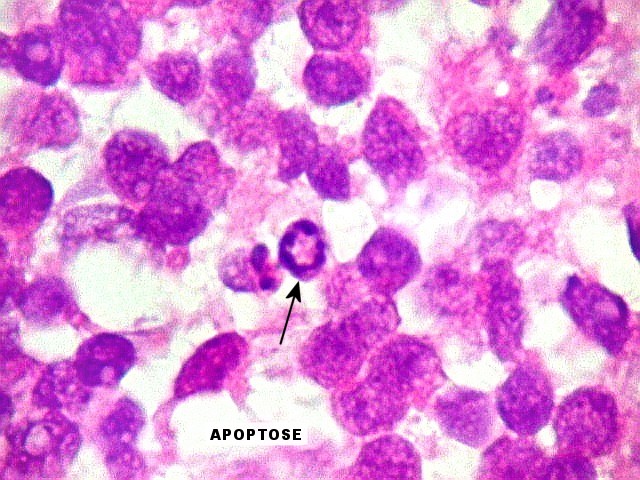
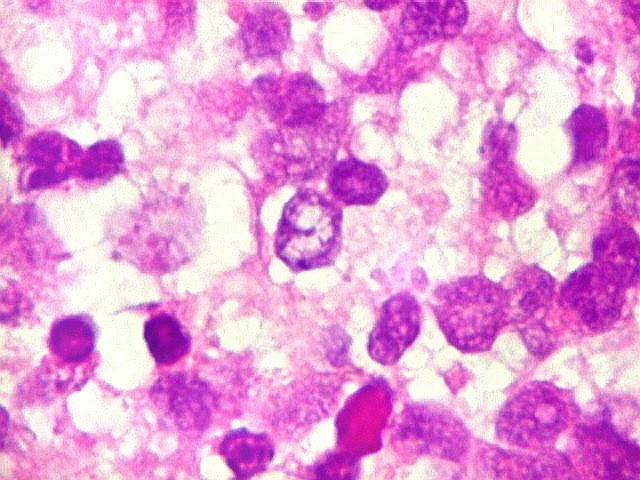
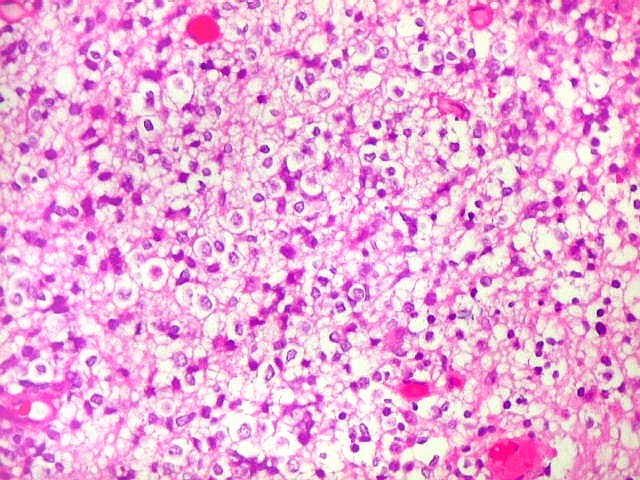
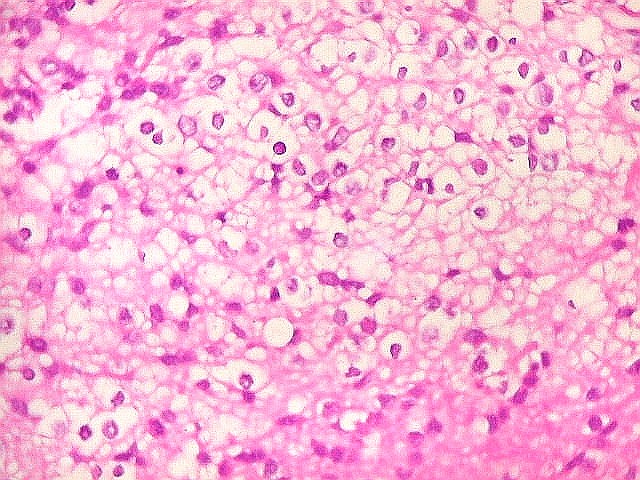
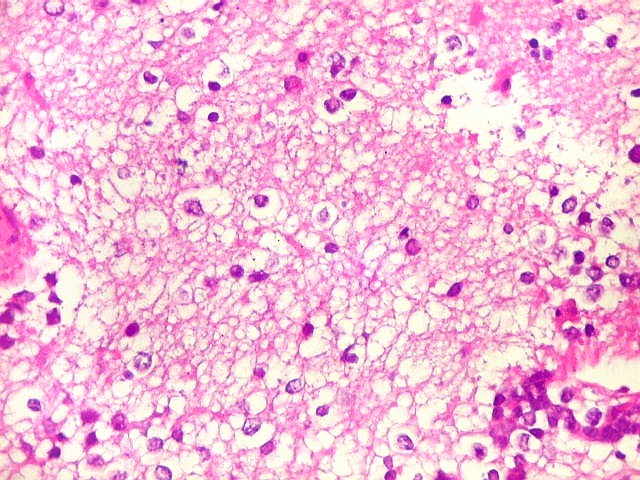
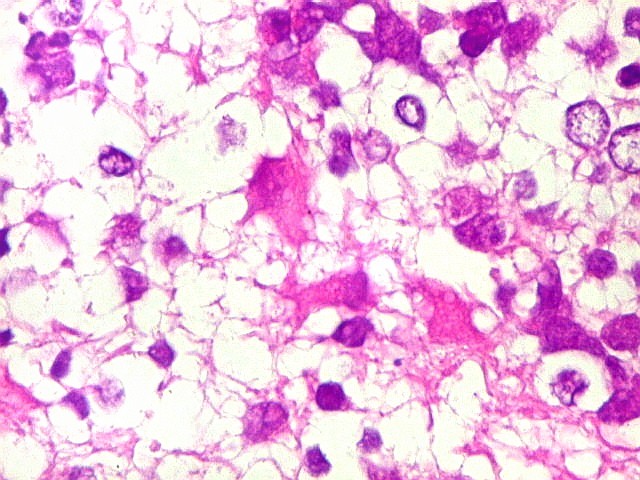
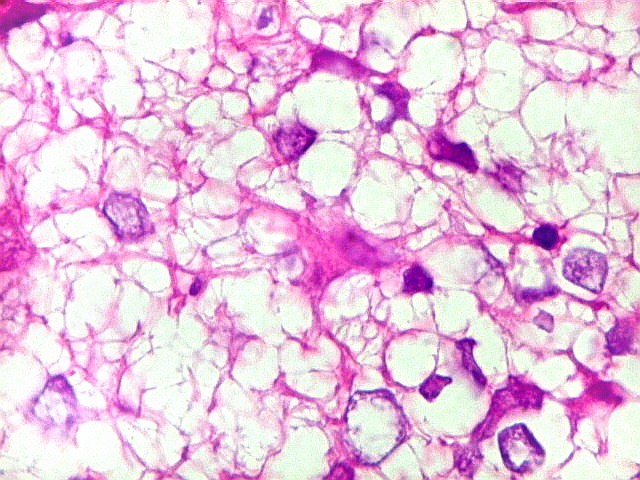
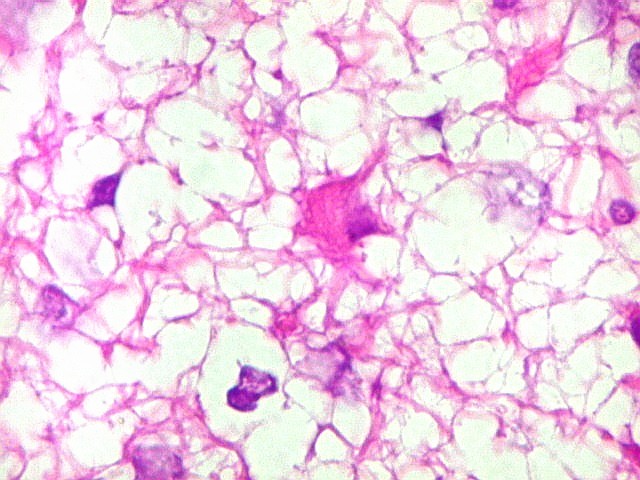
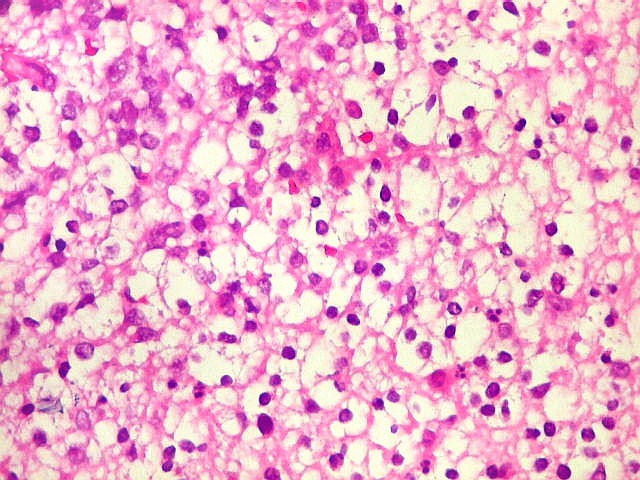
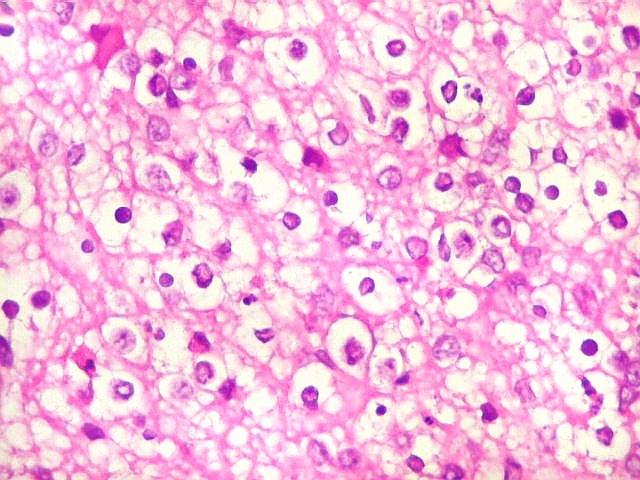
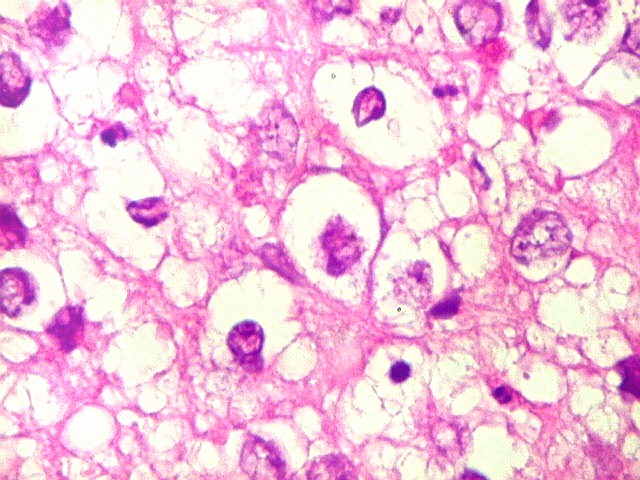
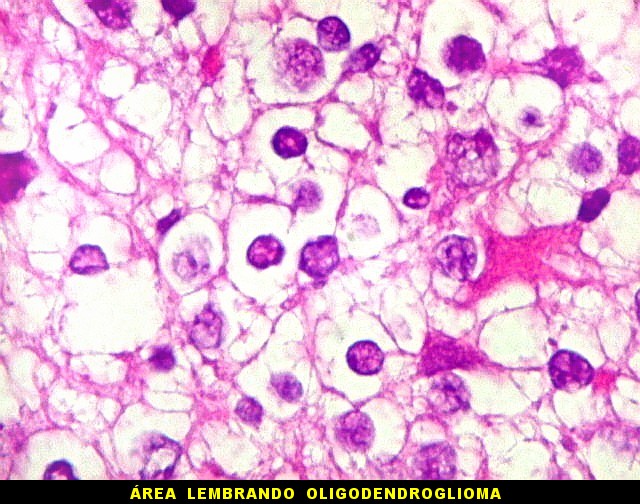
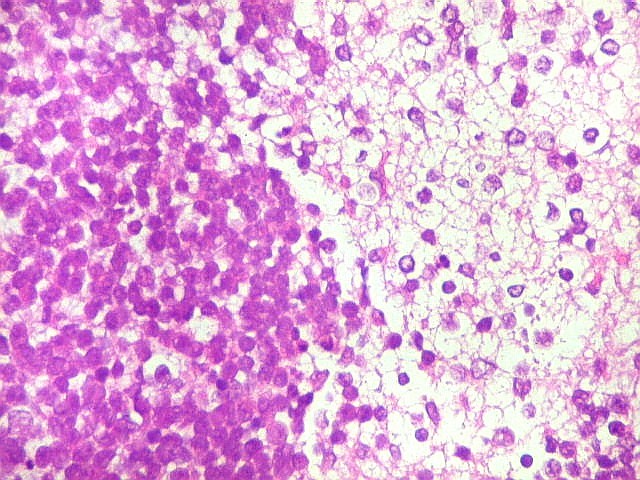
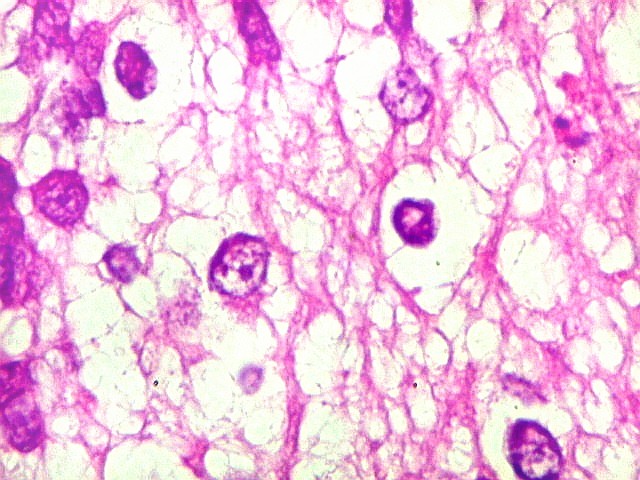
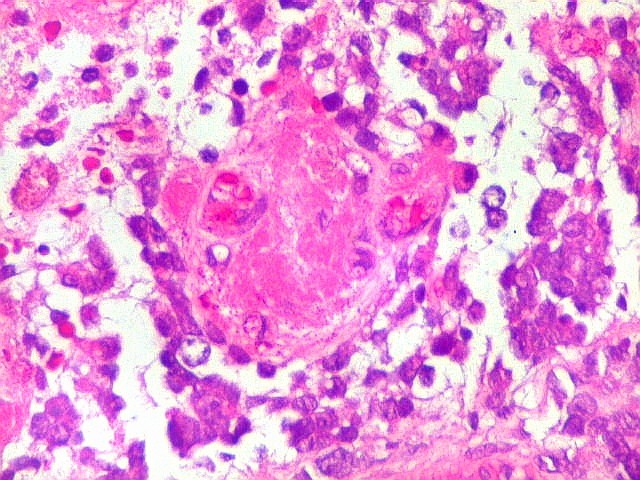
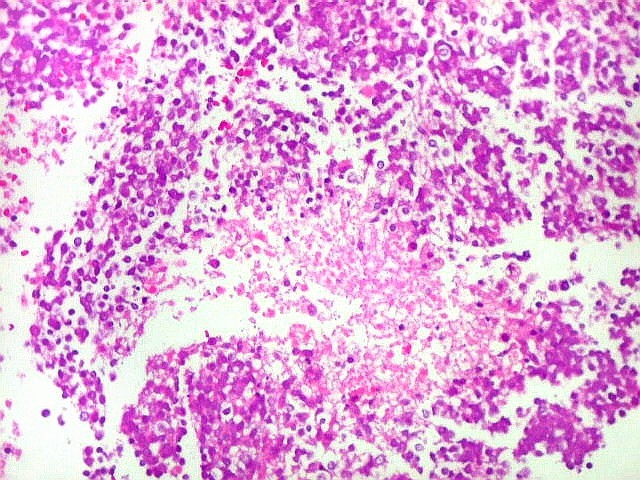
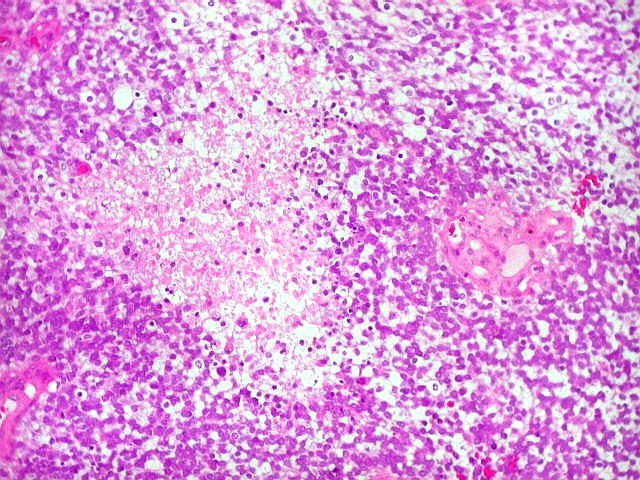
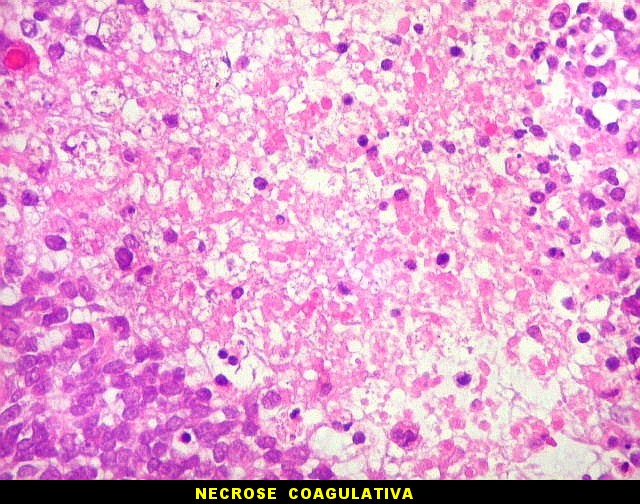
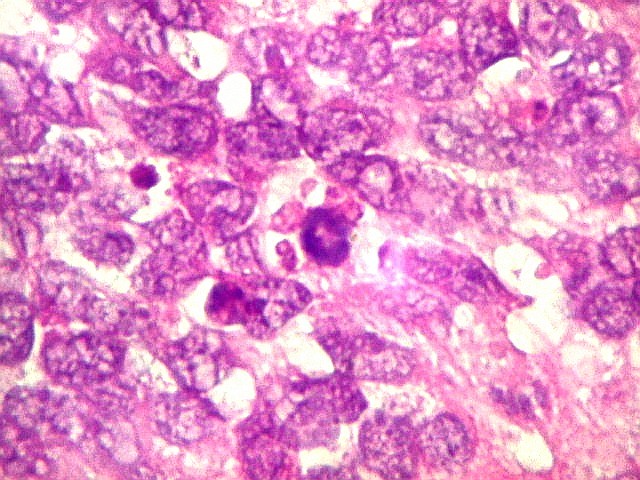
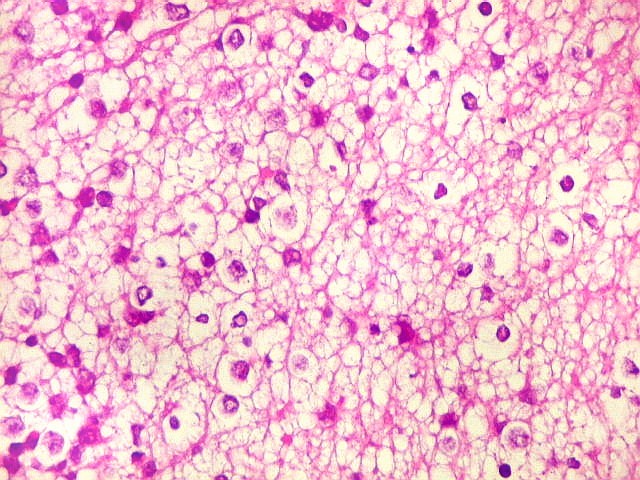
Apesar do componente
clássico de células rabdóides, essas não
são obrigatórias, sendo o tumor composto em grande parte
ou completamente de células indiferenciadas de tamanho médio
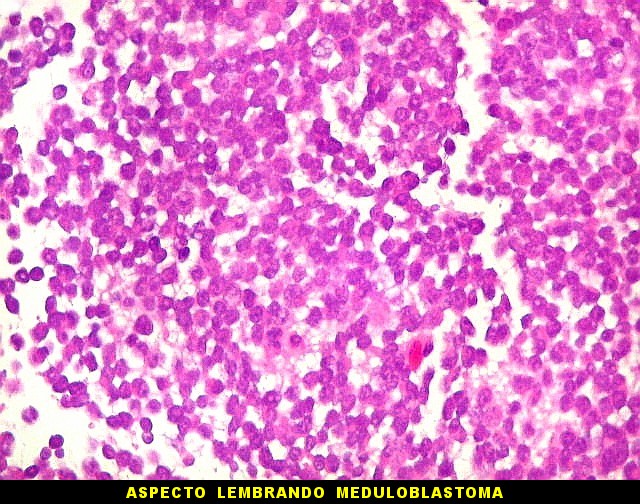
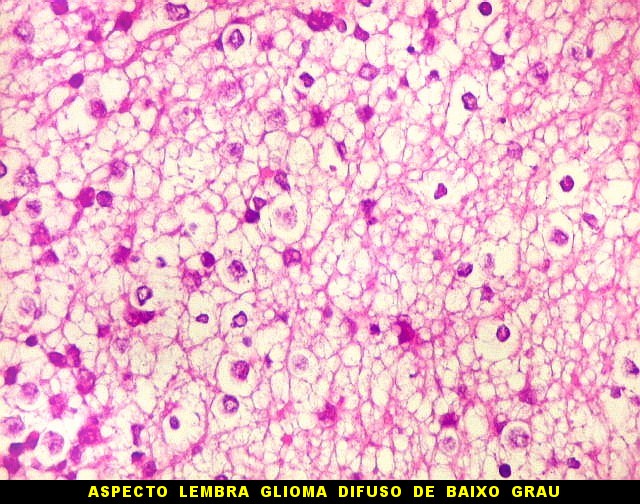
sem feições rabdóides. Levanta hipótese
de meduloblastoma ou PNET. Em tecidos moles lembra sarcoma de Ewing
ou sarcoma indiferenciado. Pode haver áreas de pequenas células
azuis.
-
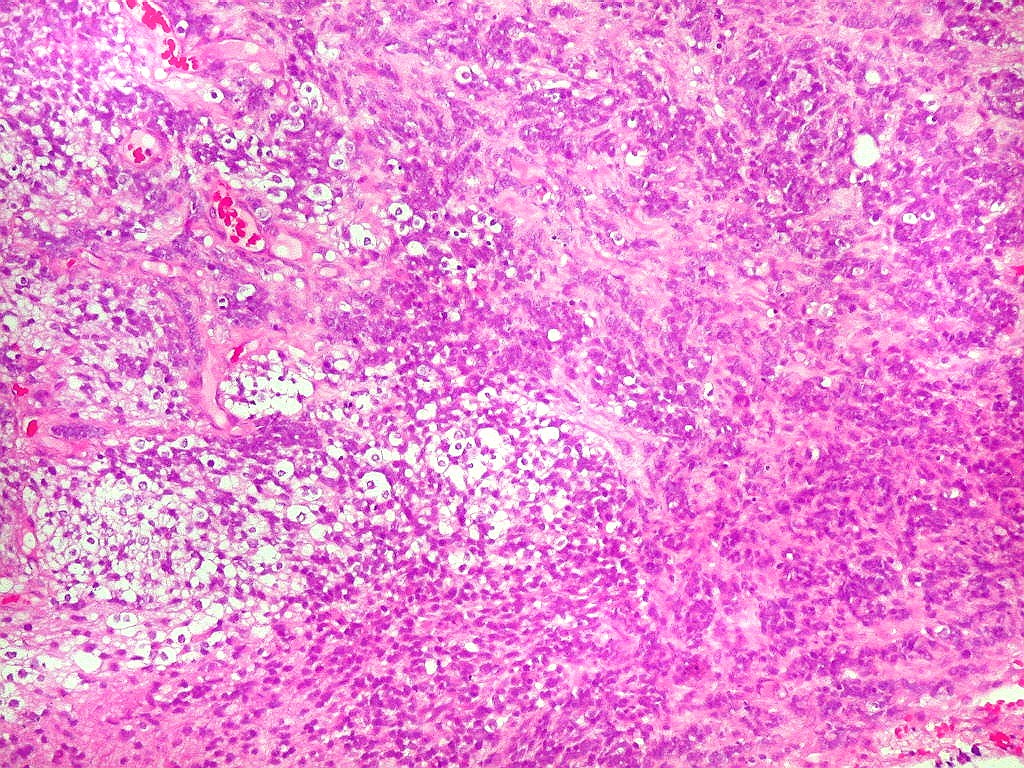
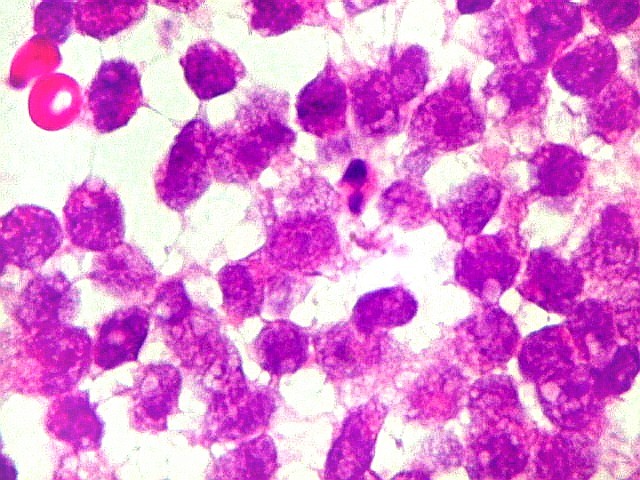
Como nos tumores
cerebrais a histologia indiferenciada não rabdóide é
relativamente comum, a pesquisa de INI-1 por imunohistoquímica é
mandatória para afastar estes diagnósticos diferenciais.
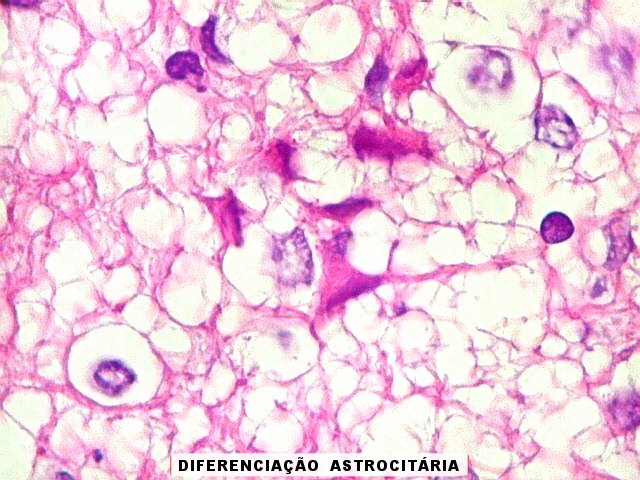
Também, o encontro de células fusiformes e estruturas epiteliais
organizadas são relativamente raras, embora enfatizadas na literatura
mais antiga. Isso levanta dúvida sobre a conveniência de continuar
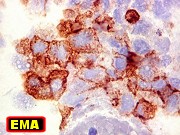
o uso do termo teratóide. A coexpressão de EMA (antígeno
epitelial de membrana) e SMA (actina de músculo liso) é relativamente
específica para ATRT.
-
Morfologia
rabdóide não relacionada a perda de INI-1 pode ser encontrada
focalmente em carcinomas, sarcomas, melanomas, tumores gliais e meníngeos
(meningioma rabdóide, OMS grau
III).
Literatura e histórico.
Referência principal.
Pawel BR. SMARCB1-deficient Tumors of Childhood: A Practical
Guide. Pediatr Dev Pathol. 2018 Jan-Feb;21(1):6-28.
Primeira
descrição de tumores rabdóides em 1978.
Beckwith
JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from
the First National Wilms Tumor Study. Cancer. 1978;41:19371948.
Primeiro
estudo ultraestrutural dos tumores rabdóides do rim.
Haas JE,
Palmer NF, Weinberg AG, Beckwith JB. Ultrastructure of malignant rhabdoid
tumor of the kidney. A distinctive renal tumor of children. Hum Pathol.
1981;12:646657.
Caracterização
dos tumores teratóides/rabdóides atípicos do sistema
nervoso central.
Rorke LB,
Packer R, Biegel J. Central nervous system atypical teratoid/rhabdoid tumors
of infancy and childhood.
J Neurooncol.
1995;24:2128.
|