|
|
|
|
|
|
|
|
| Masc. 17 a. História iniciada em julho de 2005 com episódio único de crise convulsiva. Detalhes. | |||
| RM 5/7/2005. Primeira lesão no lobo parietal esquerdo | RM 21/9/2005. Rápido crescimento em 2½ meses | 1a. amostra (2005). Sarcoma mixóide, grau histológico 2 | RM 8/3/2006. Primeira recidiva, temporal esquerda |
 |
 |
 |
 |
| RM 6/8/2008. Segunda recidiva, parietal esquerda, em local diferente da lesão original | 3a. amostra. Sarcoma indiferenciado de alto grau histológico, padrão predominante de células fusiformes. | RM 7/8/2009. Terceira recidiva, frontal esquerda. | RM 1/10/2009. Rápido crescimento em 2 meses |
 |
 |
 |
 |
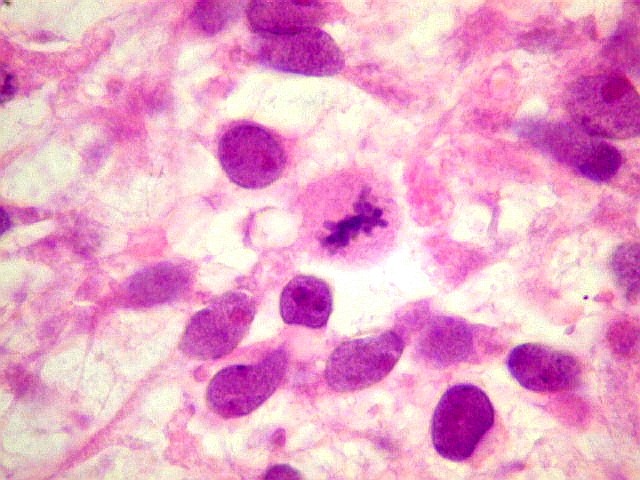
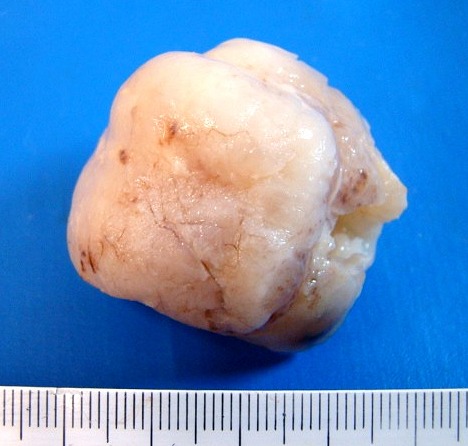
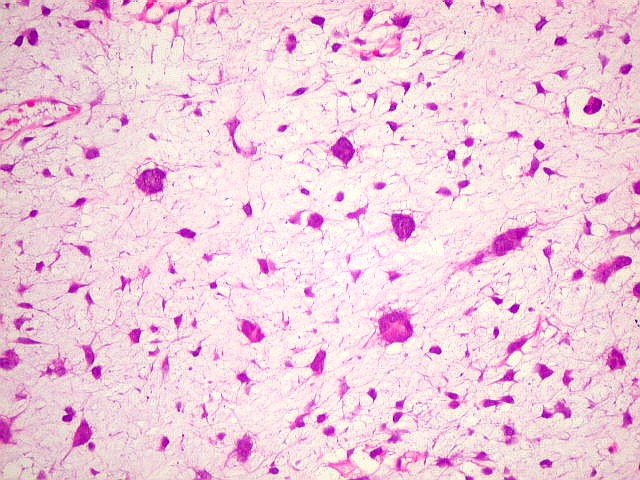
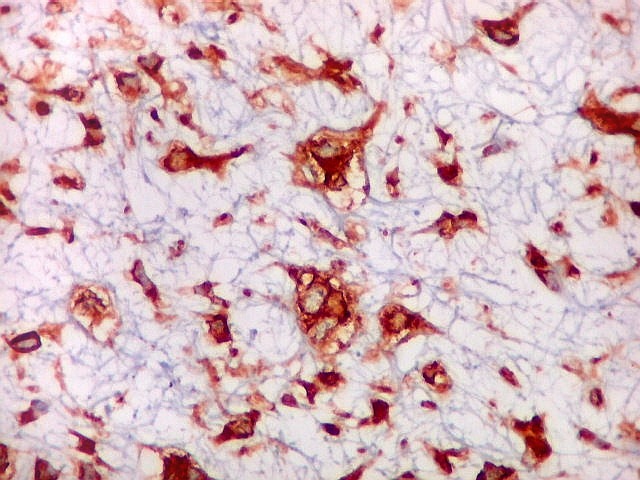
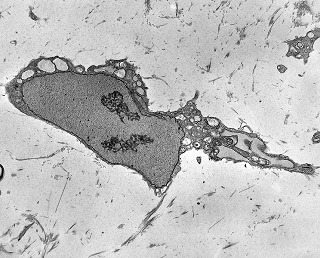
| 4a. amostra - macro | HE. Sarcoma mixóide rico em células pleomórficas | IH. Positivo para vimentina (foto), desmina, CD56, CD57, CD34. | ME. Células de padrão fibroblástico em matriz amorfa com fibrilas colágenas. |
 |
 |
 |
 |
| Conclusão - neoplasia maligna de linhagem conjuntiva (sarcoma), com padrão histológico mixóide, primário do sistema nervoso central, com recidivas múltiplas em diferentes locais. Variação do aspecto histológico e grau de malignidade nas amostras. Ver comentário e textos de apoio: sarcomas cerebrais, fibrossarcoma, mixofibrossarcoma. | |||
| Masc. 17
a.
Julho de 2005 episódio único de crise convulsiva. RM revelou lesão parietal esquerda única, que, em RM de controle após 2 meses, sofreu considerável expansão. Tumor foi retirado e o diagnóstico anátomo-patológico em outro laboratório foi astrocitoma grau II (depois revisto neste serviço como sarcoma mixóide). Não foi realizada terapia adjuvante. Março de 2006 recidiva como lesão temporal esquerda, distante da primeira cirurgia (para RM, clique). Ressecada sem intercorrências. AP original - astrocitoma grau III (posteriormente revisado como sarcoma, não apresentado aqui). Fez radio- e quimioterapia, com remissão total nos exames de controle. Agosto 2008 convulsões de difícil controle. RM segunda recidiva, em região diferente das anteriores. Extirpação total da porção visível. AP original - astrocitoma grau III (também revisto neste serviço como sarcoma indiferenciado de alto grau histológico). Agosto 2009 RM de controle mostrou nova lesão, agora frontal E, em local diferente de todas anteriores, com rápido crescimento em 2 meses. Nova cirurgia, a quarta, em fins de setembro de 2009. Diagnóstico AP neste serviço - sarcoma mixóide. Clique para espécime macro, microscopia em HE, colorações especiais, imunohistoquímica e microscopia eletrônica. Em todas cirurgias a lesão apresentava bom plano de clivagem do tecido cerebral normal, com características às vezes sarcomatosas às vezes gelatinosas (sic), pouco sangrante, de cor púrpura, geralmente saindo em bloco. Todas cirurgias tiveram evolução pós-operatória adequada, com mínimas seqüelas. |
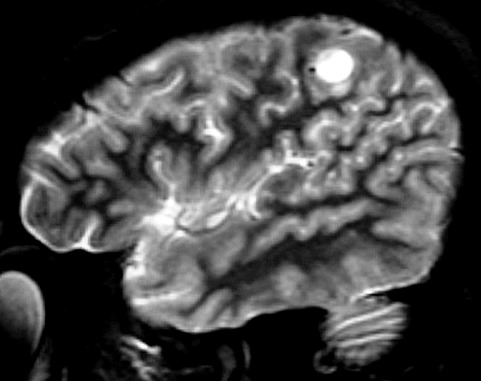
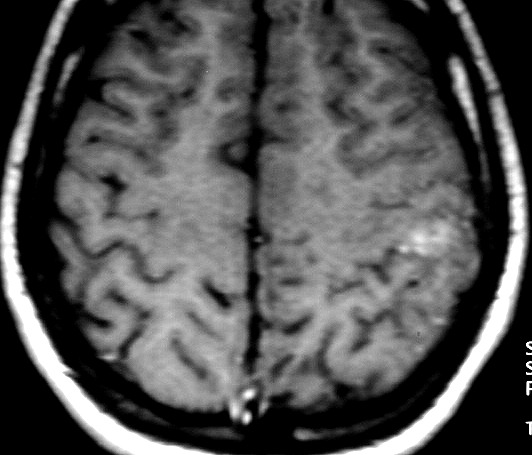
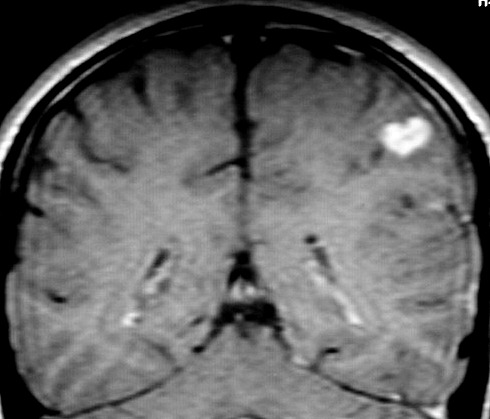
| RESSONÂNCIA MAGNÉTICA 5/7/2005 - MELHORES CORTES. Pequena e única lesão superficial no lobo parietal E, envolvendo córtex e substância branca. Lesão é bem delimitada, de contorno ovalado ou levemente irregular e, em alguns cortes, parece ter-se originado em um sulco cerebral. Não se acompanha de edema perilesional nem efeito de massa. Mostra hipersinal em T2 e FLAIR. Com contraste, parece haver impregnação (não há imagens sem contraste para comparar). | ||
| AXIAL,_T1 COM CONTRASTE | CORONAL, T1 COM CONTRASTE | SAGITAL, T2 |
 |
 |
|
| Este exame em detalhe. | ||
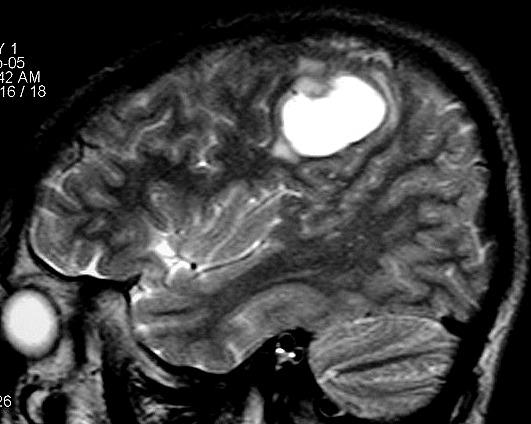
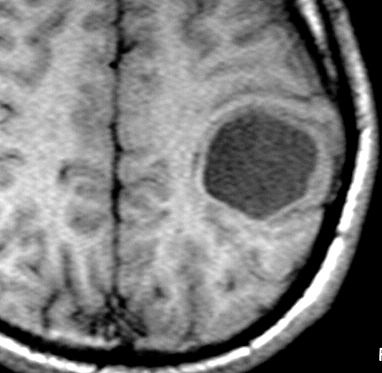
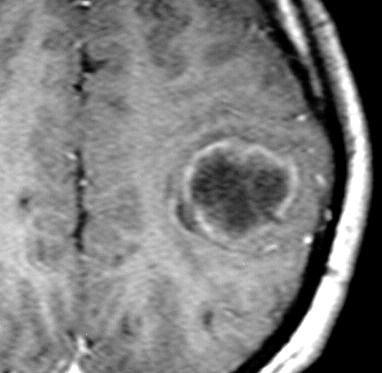
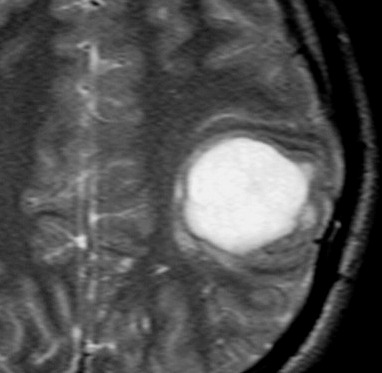
| RM, 21/9/2005 - AXIAIS. Neste segundo exame pré-operatório, 2 meses e meio após o primeiro, houve grande expansão da lesão, que mantém boa delimitação, discretos efeito de massa e edema perilesional. O tumor apresenta hipossinal em T1, hipersinal em T2 e FLAIR e impregnação periférica por contraste. | ||
| T1 SEM CONTRASTE | T1 COM CONTRASTE | T2 |
 |
 |
 |
| AXIAL, FLAIR | CORONAL, T1 COM CONTRASTE | SAGITAL, T2 |
 |
 |
|
| Este exame em detalhe. | ||
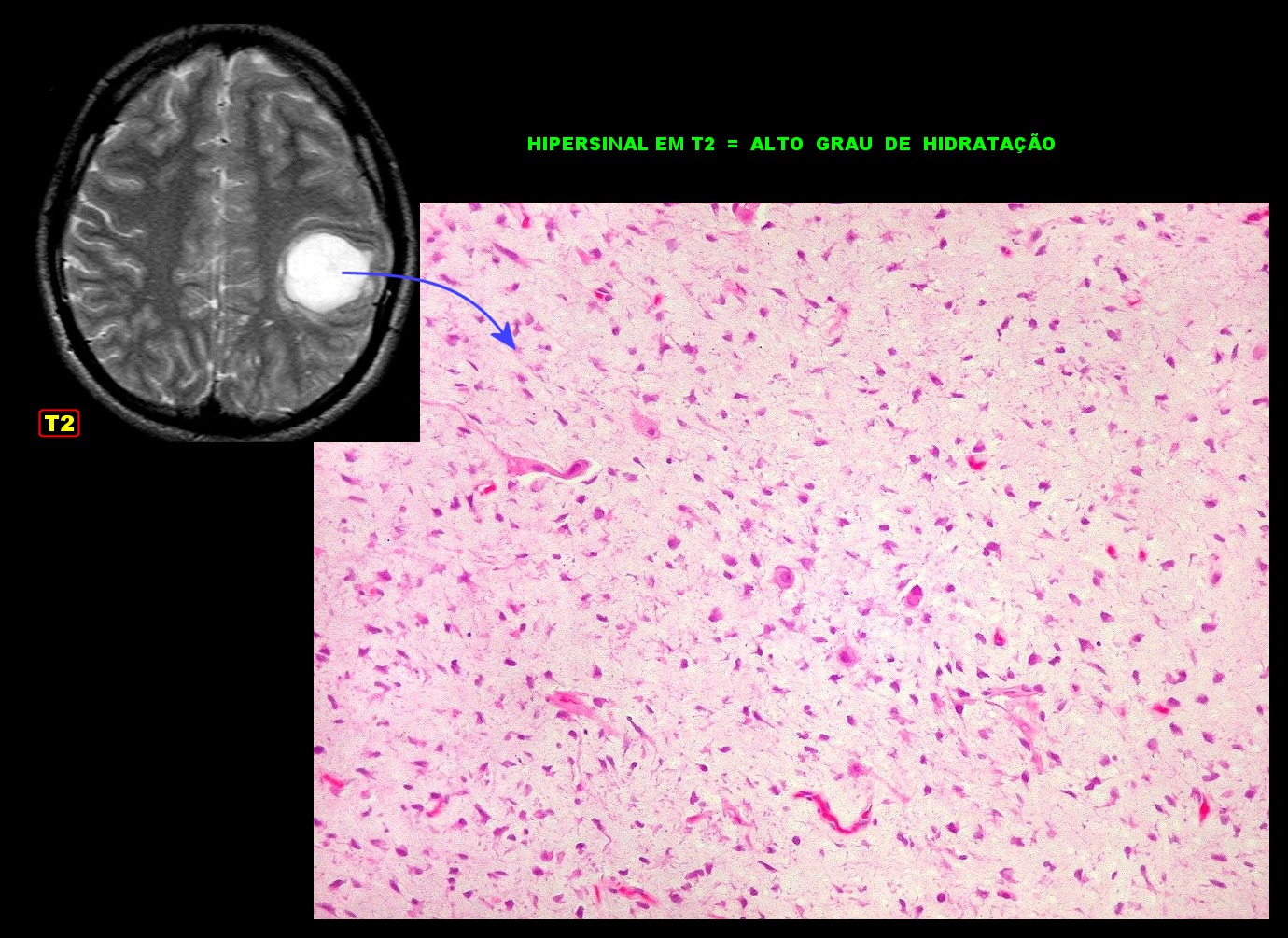
| Primeira
amostra (2005). Correlação com imagem.
O alto grau de hidratação do tecido, devido à abundante matriz mixóide entre as células neoplásicas, correlaciona-se com o hipersinal em T2. Mais imagens deste exame e da lâmina. |
 |
| Primeira amostra (2005). Destaques da microscopia. Clique para exame em detalhe. | ||
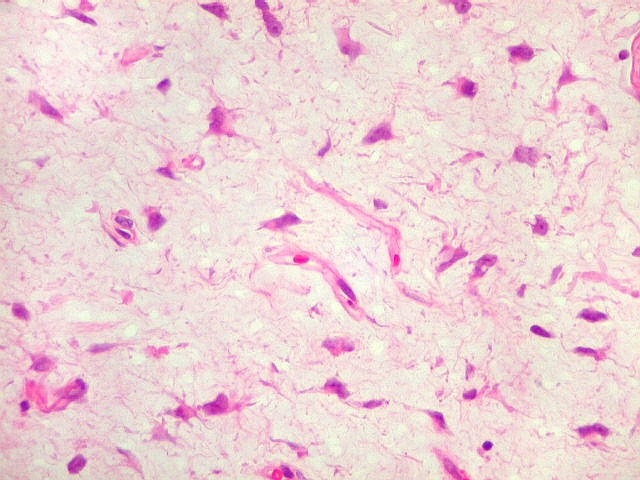
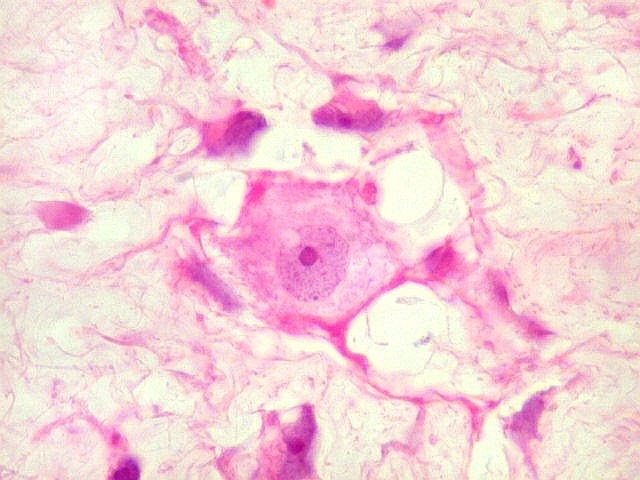
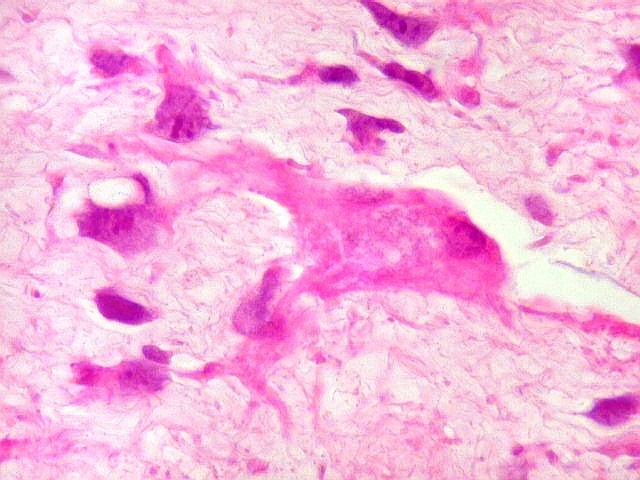
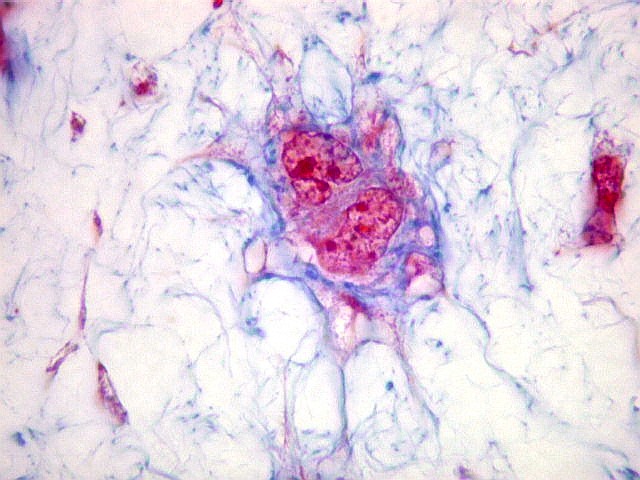
| HE. Caráter sarcomatoso mixóide, com células estreladas e celularidade variável, em abundante matriz extracelular. | Células lembrando neurônios. | Células lembrando astrócitos |
|
 |
 |
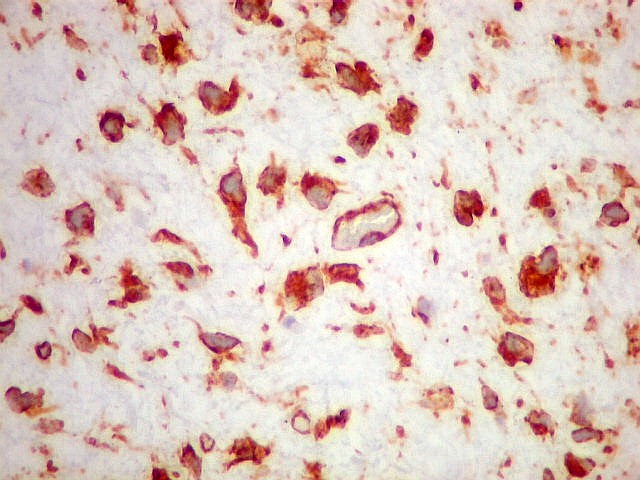
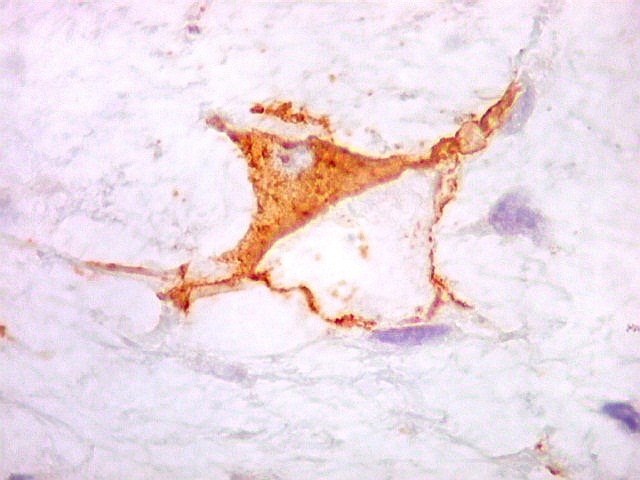
| VIM. Positividade nas células neoplásicas e em vasos. | GFAP. Positivo em astrócitos pré-existentes, negativo no tumor. | Ki-67. Positivo em raros núcleos de células neoplásicas. |
 |
 |
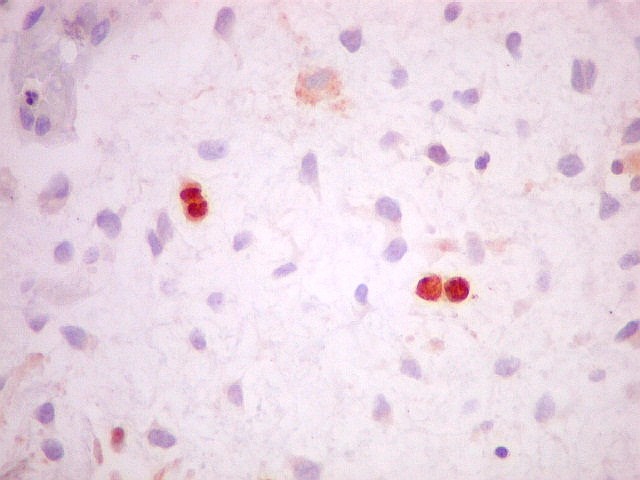 |
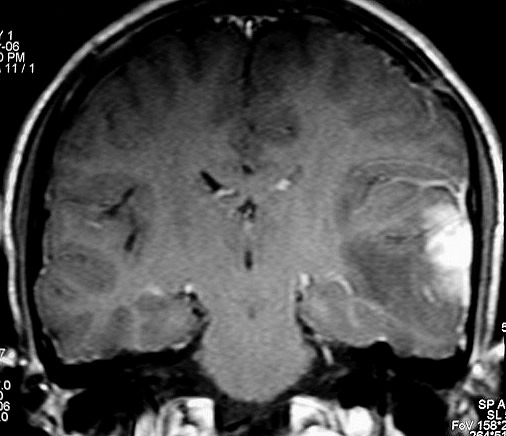
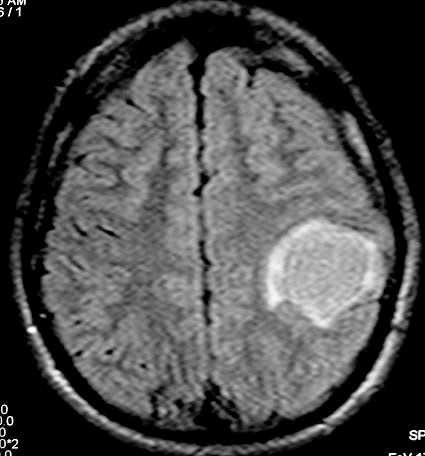
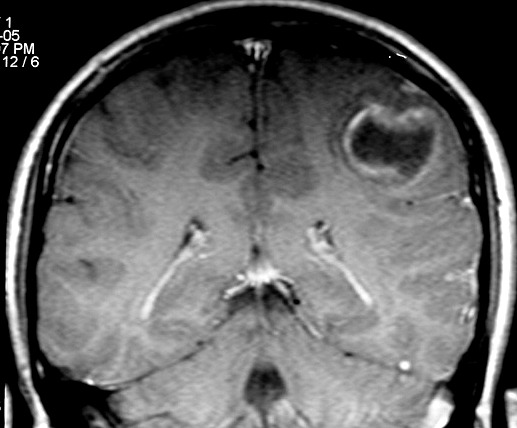
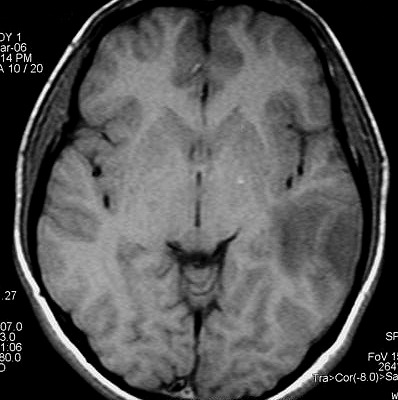
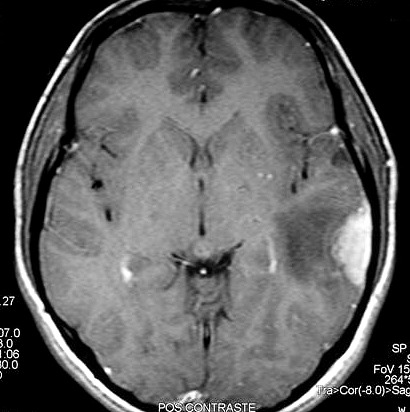
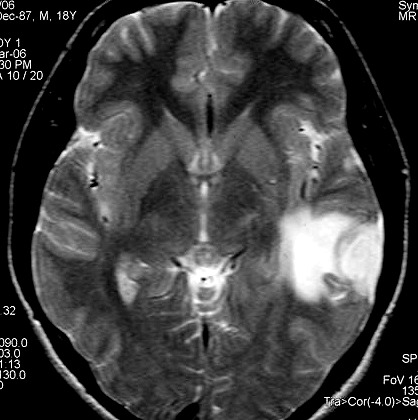
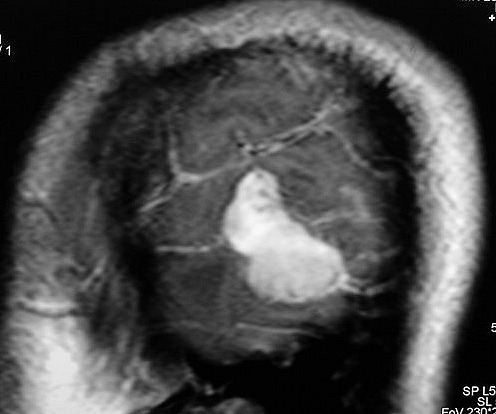
| RM 8/3/2006 - Após ressecção do tumor inicial (parietal E, acima) em 10/2005, foi detectada outra lesão em 3/2006, em local distante da primeira, no lobo temporal esquerdo, mais superficial, aderida à dura-máter, com aspecto em calota. Apresentava hipossinal em T1, hipersinal em T2, forte impregnação homogênea e edema perilesional. O aspecto histológico foi semelhante ao da primeira amostra (não demonstrado aqui). Fez RT, QT, ficou assintomático. | ||
| AXIAIS, T1 SEM CONTRASTE | T1 COM CONTRASTE | T2 |
 |
 |
 |
| CORONAL, SAGITAL, T1 COM CONTRASTE | SAGITAL, T2 | |
|
 |
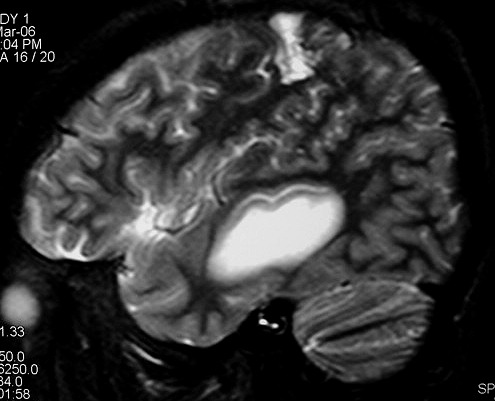 |
| Este exame em detalhe. | ||
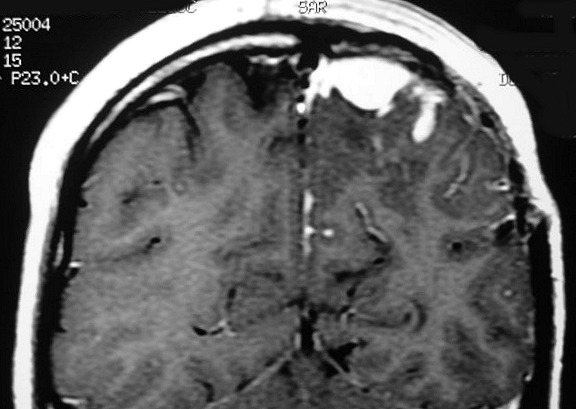
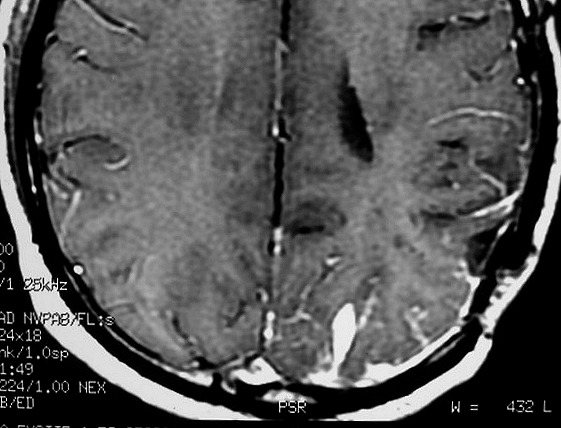
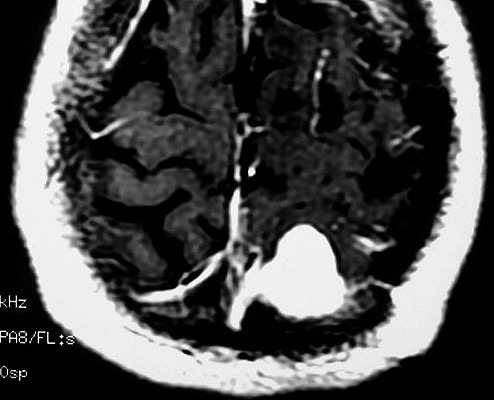
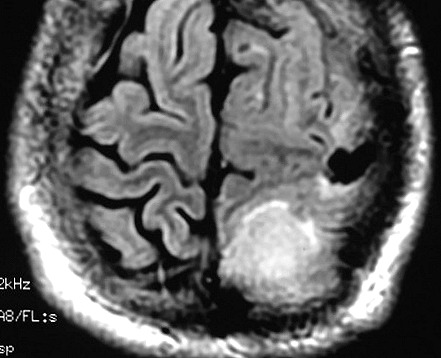
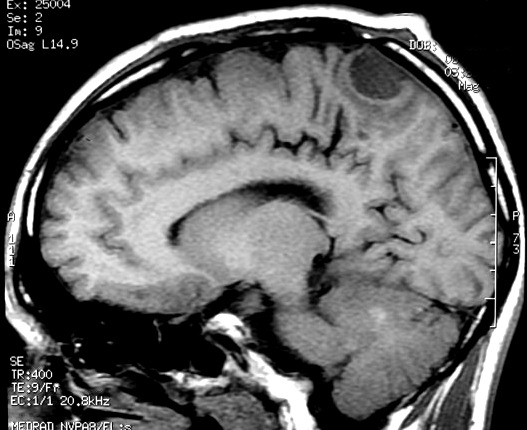
| RM 6/8/2008 - Terceira lesão detectada devido a convulsões de difícil controle, 3 anos após início da história. Surgiu em local diferente das anteriores, no lobo parietal E, mais medial e posterior que a lacuna da primeira cirurgia. As características de imagem foram semelhantes às da primeira e segunda lesões. Além de um nódulo captante bem delimitado, com hipossinal em T1 e hipersinal em T2 e FLAIR, havia áreas adjacentes de infiltração meníngea, em que a lesão preenchia sulcos superficiais parietais E. Lesão foi extirpada cirurgicamente. O anátomo-patológico mostrou aumento no grau de malignidade. RM de controle após 6 meses não revelou resíduos captantes. | ||
| AXIAIS, T1 COM CONTRASTE | FLAIR | |
 |
 |
 |
| CORONAL, T1 COM CONTRASTE | SAGITAL, T1 SEM CONTRASTE |
|
 |
| Este exame em detalhe. | |
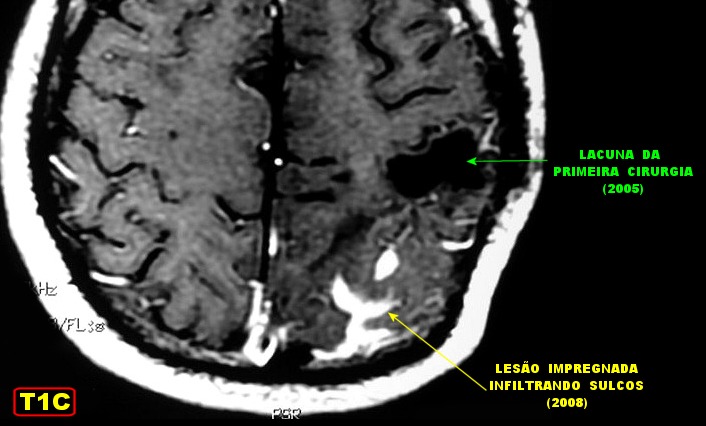 |
| Terceira amostra (2008). Destaques da microscopia. Clique para exame em detalhe. | ||
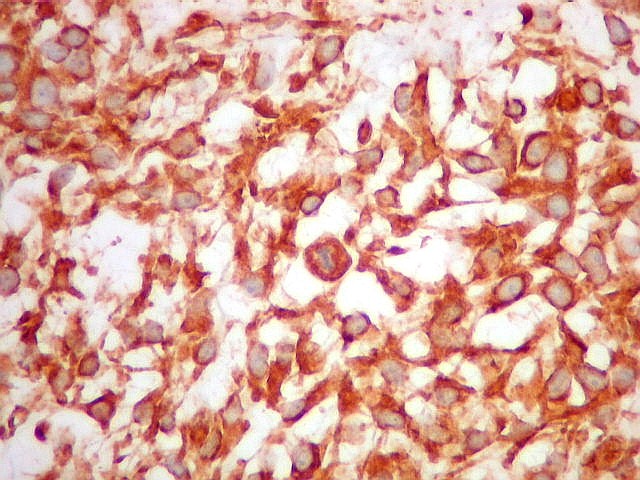
| HE. Neoplasia fusocelular de alta celularidade, rica em mitoses. | VIM. Positividade citoplasmática difusa e intensa. | Ki-67. Positividade em cerca de 10 a 15% dos núcleos das células neoplásicas |
|
 |
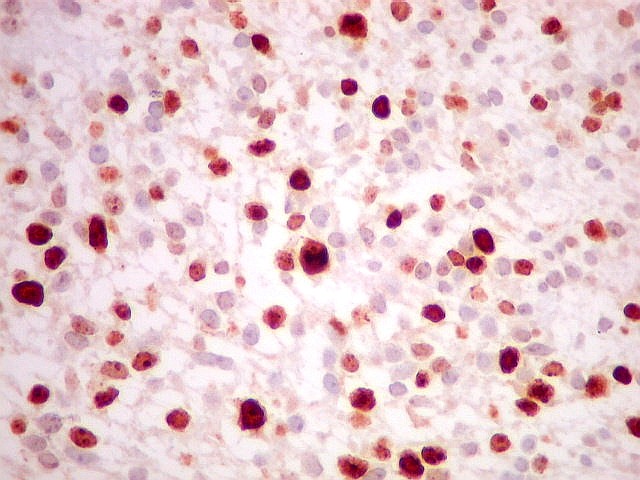 |
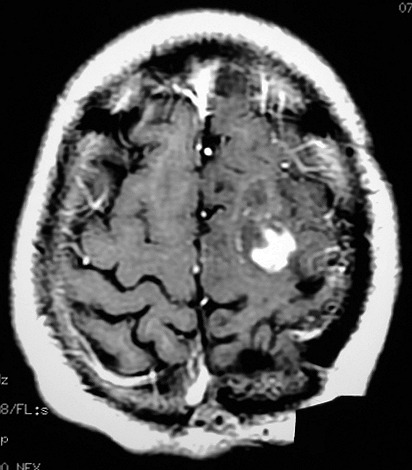
| RM 7/8/2009 - T1 COM CONTRASTE. Quarta lesão, 4 anos após o início da história, agora nos giros frontal superior e pré-central E, próxima da linha média. Características semelhantes às das anteriores. Em 50 dias o tumor expandiu-se consideravelmente (abaixo). | ||
|
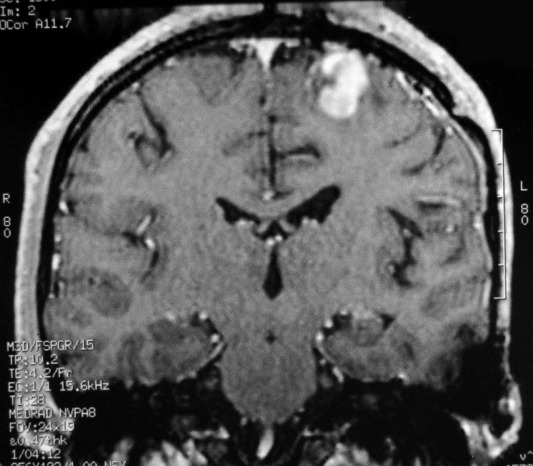 |
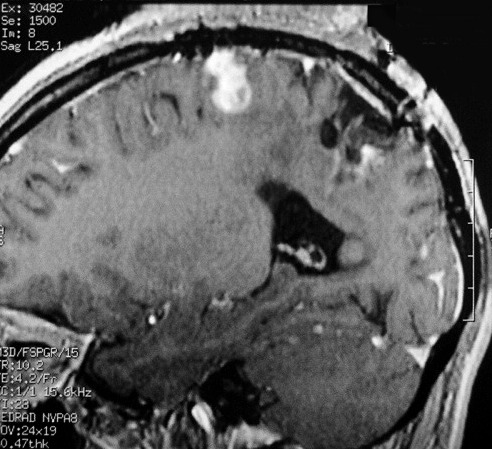 |
| Mais imagens deste exame. | ||
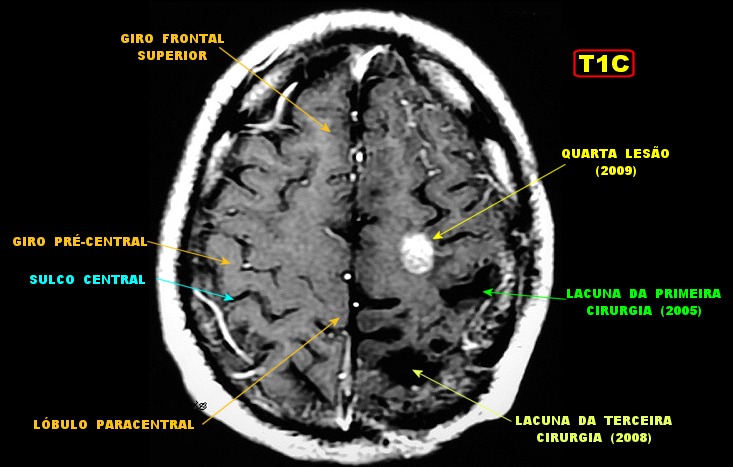 |
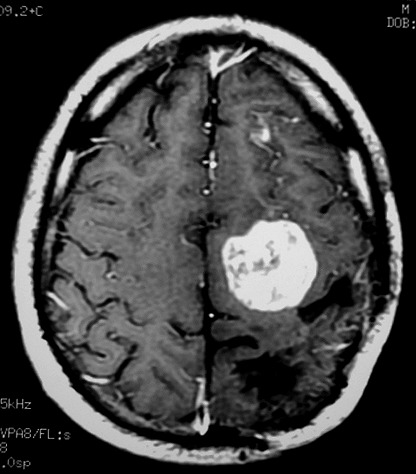
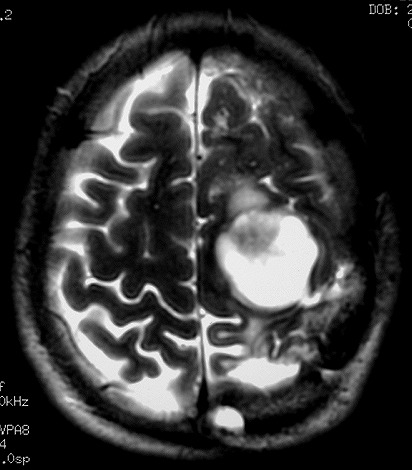
| RM 1/10/2009 - Em menos de 2 meses a lesão cresceu aproximadamente 4 vezes em diâmetro (portanto, presumivelmente, cerca de 60 vezes em volume, pois 4³=64), recapitulando o ocorrido quando da primeira lesão (RMs de 5/7/2005 e 21/9/2005). | ||
| AXIAIS, T1 SEM CONTRASTE | T1 COM CONTRASTE | T2 |
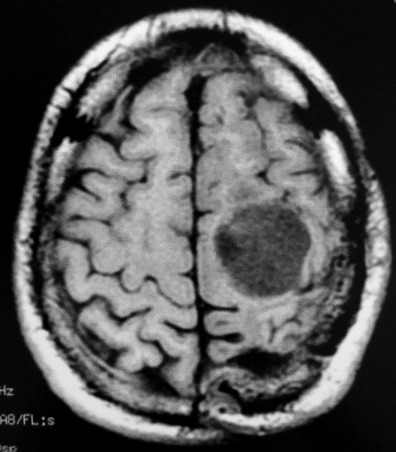 |
|
 |
| AXIAL, FLAIR | CORONAL, T1 COM CONTRASTE | SAGITAL, T1 SEM CONTRASTE |
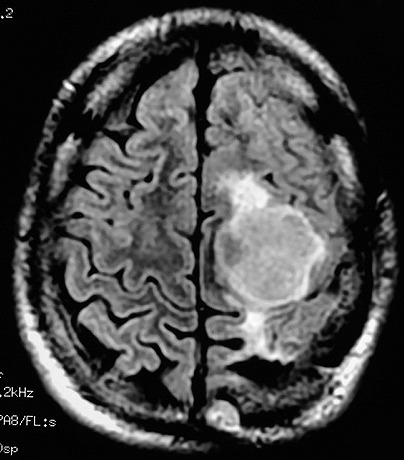 |
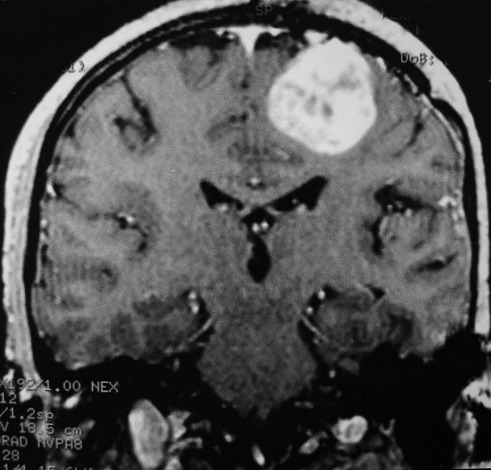 |
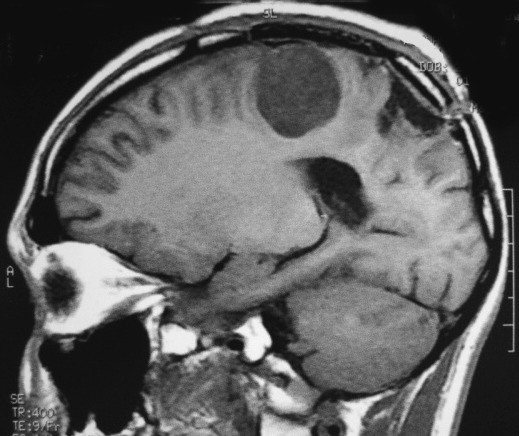 |
| Este exame em detalhe. | ||
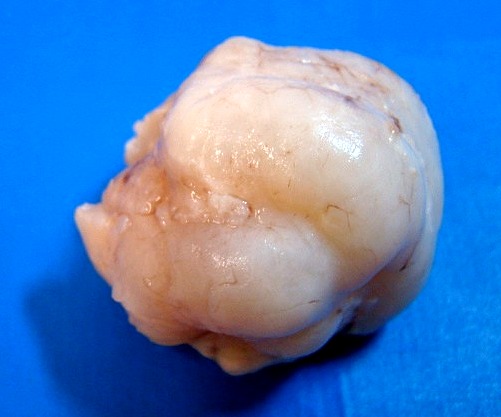
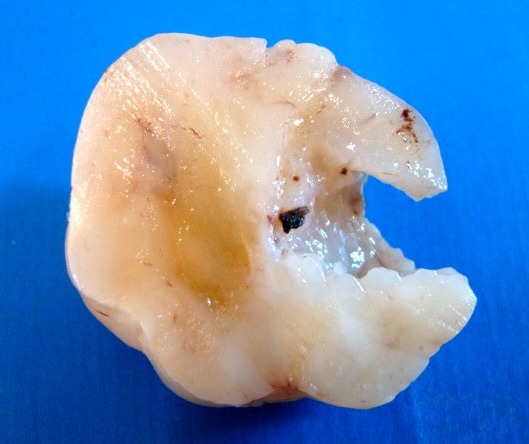
| Quarta amostra (2009). Macroscopia da peça. Mais imagens. | ||
|
 |
 |
| Destaques da microscopia e imunohistoquímica. | ||
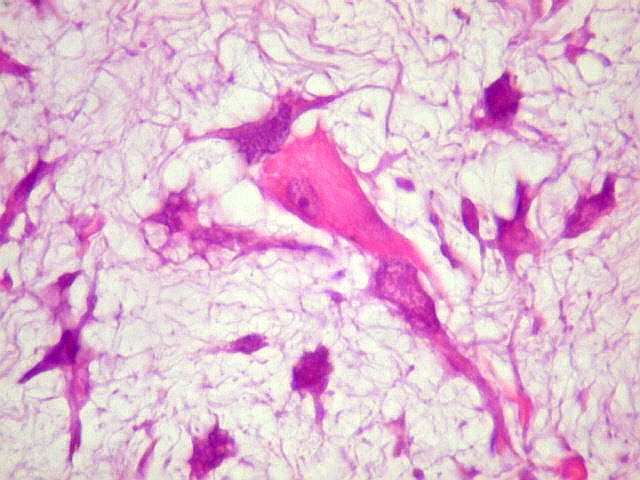
| HE. Células fusiformes, estreladas e pleomórficas, muitas multinucleadas, em abundante material intersticial mixóide | Astrócitos aprisionados pela neoplasia evidenciam caráter infiltrativo | Masson. Fibrilas colágenas se destacam na substância fundamental amorfa |
|
 |
 |
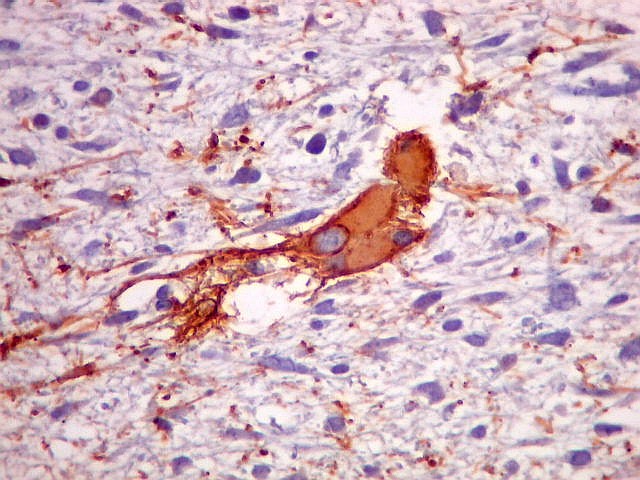
| VIM. Positividade citoplasmática difusa e universal nas células tumorais | GFAP. Positivo em astrócitos pré-existentes englobados pelo tumor, negativo nas células neoplásicas | Desmina. Positividade citoplasmática nas células neoplásicas, inclusive em mitoses |
|
 |
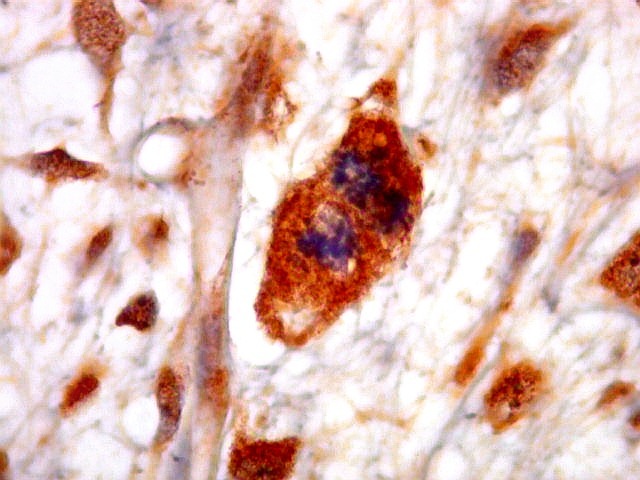 |
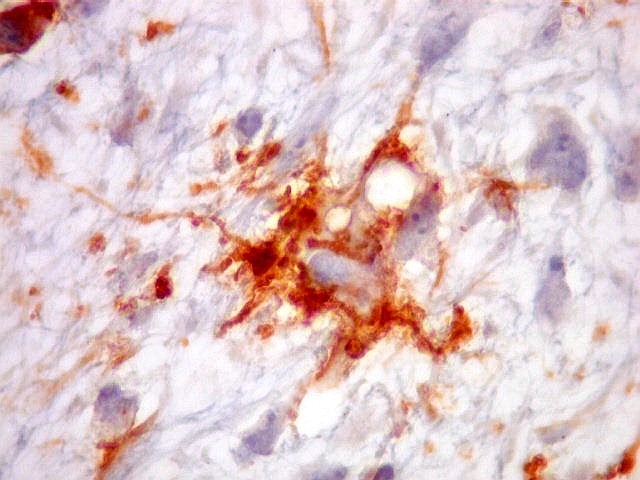
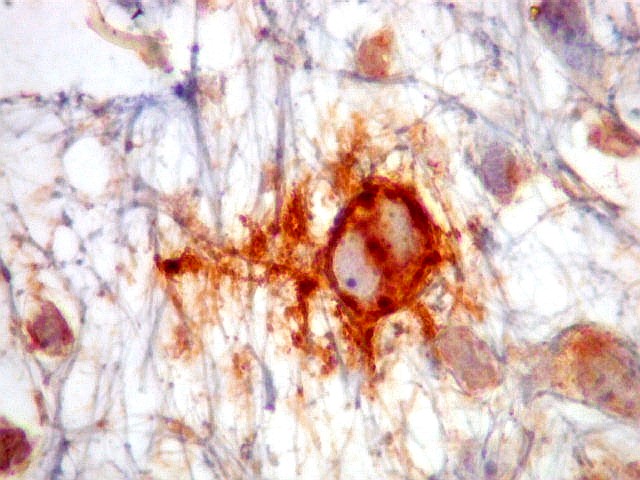
| HHF-35. Positivo em muitas células neoplásicas 1A4 - negativo | CD56. Positividade citoplasmática em parte das células neoplásicas | CD57. Idem |
 |
 |
 |
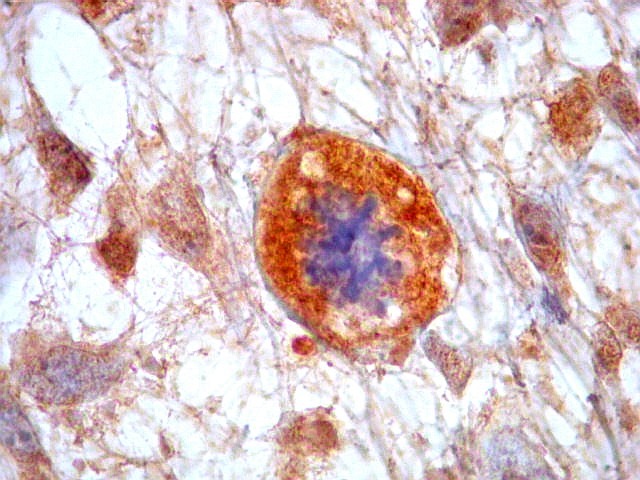
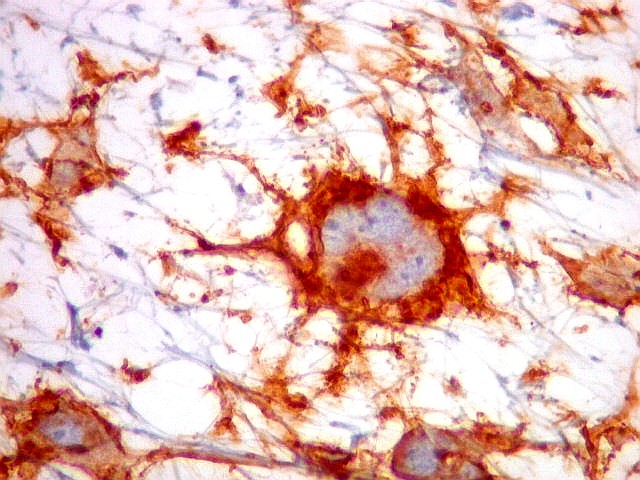
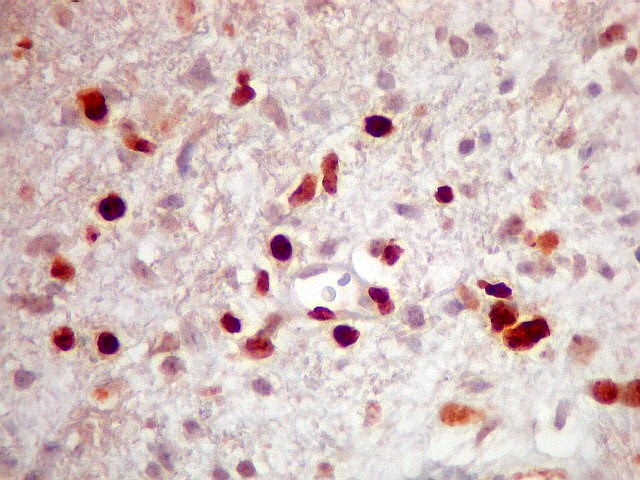
| CD34. Positividade citoplasmática em parte das células neoplásicas e em vasos | Ki-67. Positividade em cerca de 10 a 15% dos núcleos das células neoplásicas | p53. Positividade em cerca de 40% dos núcleos das células neoplásicas |
 |
 |
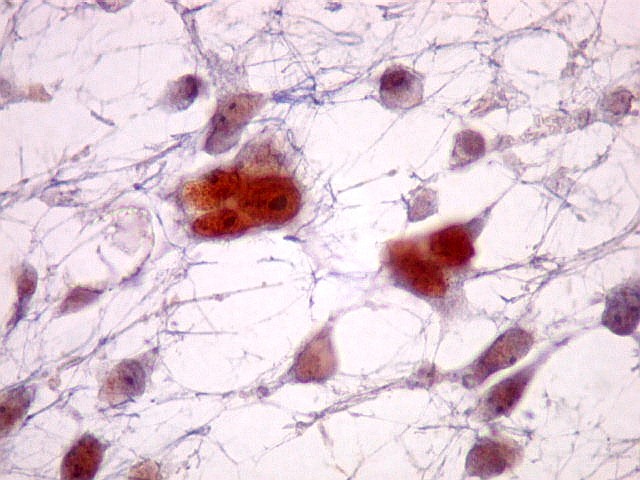 |
| Destaques da microscopia eletrônica. | ||
| Células neoplásicas. | Organelas. Abundante retículo endoplasmático rugoso. | Interstício. Fibras colágenas, substância fundamental amorfa. |
|
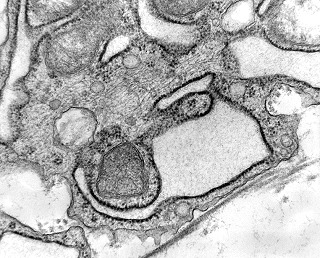 |
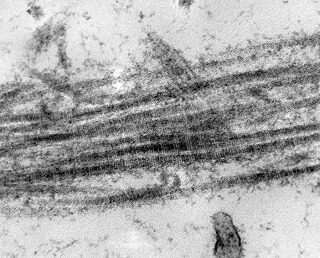 |
| Vasos. | Axônios mielínicos seqüestrados pelo tumor. | Astrócitos reativos, de padrão gemistocítico |
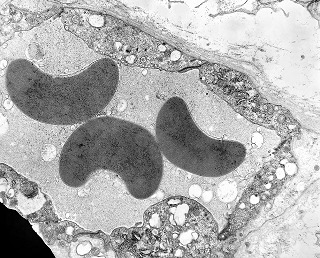 |
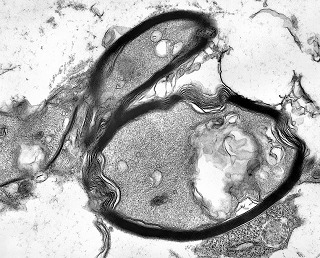 |
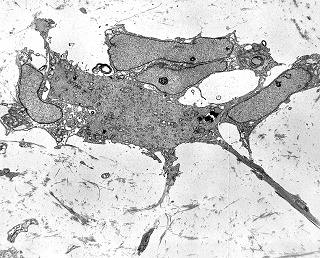 |
| Comentário.
Dificuldade diagnóstica, aliviada pela correlação com neuroimagem. Este caso foi selecionado para apresentação detalhada porque ilustra de forma eloqüente a importância da correlação entre neuroimagem e neuropatologia para um correto diagnóstico. As duas abordagens (imagenológica e anátomo-patológica) podem ser comparadas às duas faces de uma mesma moeda. A imagem mostra a macroscopia da lesão com o paciente intacto, informando dados da maior relevância, como topografia, tamanho e número de lesões, delimitação, relação com estruturas vizinhas, e feições intrínsecas do tecido, como homogeneidade, grau de hidratação, e status da barreira hemoencefálica, através da impregnação por contraste. São dados inestimáveis, a serem cotejados com o aspecto histológico. Este pode ser de difícil interpretação per se, já que tumores de linhagens diferentes podem assemelhar-se e causar confusão. A comparação com as imagens minora esta possibilidade e estabelece bases firmes para um diagnóstico consistente, tão próximo da verdade quanto possível. No presente caso, o espécime histológico inicial consistia de neoplasia de células fusiformes ou estreladas, com prolongamentos citoplasmáticos de limites imprecisos, dispersas em matriz frouxa, amorfa e levemente basófila. Não havia mitoses, proliferação vascular ou necrose. A semelhança com um astrocitoma difuso de baixo grau de padrão protoplasmático era, portanto, notável. Também, a localização em hemisfério cerebral apontava nesta direção, e o diagnóstico seria compatível com a idade do paciente. Contudo, a RM inicial mostrava lesão nodular bem delimitada e captante de contraste, duas feições impróprias para astrocitomas grau II, que são difusos, mal delimitados e não se impregnam. A segunda RM, após 2 meses e meio e antes da primeira cirurgia, revelou aumento explosivo da lesão, com impregnação periférica, dados também em discordância com a hipótese de um tumor glial de baixo grau histológico. A análise dos vários espécimes por HE, colorações especiais, imunohistoquímica e (no último) microscopia eletrônica, demonstrou que as células neoplásicas, apesar de imitarem astrócitos em HE, eram GFAP negativas, contrariando uma vez mais a hipótese de astrocitoma. Havia apenas alguns astrócitos pré-existentes, aprisionados pelo crescimento e invasão do tumor. As fibrilas eosinófilas no meio da matriz amorfa, que sugeriam prolongamentos astrocitários, eram, na realidade, colágeno, como notado com o tricrômico de Masson e em ME. Com isso, o diagnóstico mais provável foi o de um raro sarcoma mixóide primário do sistema nervoso central, que poderíamos classificar, do ponto de vista estritamente morfológico, como mixofibrossarcoma. A origem do mesmo não é clara. Propomos que tenha-se iniciado na leptomeninge ou em vasos. Imagens da primeira RM, quando o tumor ainda tinha cerca de 1 cm, sugerem que se situasse em um sulco na região parietal esquerda. |
| Sarcomas
cerebrais.
Tumores cerebrais mesenquimais não meningoteliais são, em conjunto, muito raros. Podem ser benignos ou malignos, variando de grau I a IV e, histologicamente, correspondem a tumores de tecidos moles ou de osso. Termos nosológicos descritivos, como sarcoma fusocelular, sarcoma de células estreladas ou polimórficas, e mixossarcoma, devem ser substituídos por designações indicativas de linhagens específicas de diferenciação, quando possível. Também deve-se evitar o termo sarcoma meníngeo, que se presta a confusão entre os meningiomas malignos e os vários tipos de sarcoma. A classificação da OMS de 2007 inclui os seguintes tumores cerebrais mesenquimais não meningoteliais: lipoma, lipossarcoma, angiolipoma, hibernoma, tumor fibroso solitário, fibrossarcoma, fibrohistiocitoma maligno (MFH), leiomioma, leiomiossarcoma, rabdomioma, rabdomiossarcoma, condroma, osteoma, osteocondroma, condrossarcoma, osteossarcoma, hemangioma, hemangioendotelioma epitelióide, angiossarcoma, sarcoma de Kaposi, sarcoma de Ewing / tumor neuroectodérmico primitivo periférico (pPNET). Dados gerais. Incidência. As várias formas de lipoma representam cerca de 0,4% dos tumores intracranianos. Os sarcomas em geral são raros. Baseado em duas séries de 19 e 17 casos, os sarcomas representaram menos de 0,1 e 0,2 % dos tumores intracranianos. Os tipos mais comuns incluem o fibrossarcoma, fibrohistiocitoma maligno e sarcomas indiferenciados. Podem ocorrer em qualquer idade, com os rabdomiossarcomas preferencialmente em crianças e o fibrohistiocitoma maligno e o condrossarcoma em adultos. Não há predileção por sexo. Causa. Fibrossarcoma, fibrohistiocitoma maligno, condrossarcoma e osteossarcoma podem ocorrer vários anos após irradiação do crânio, mais comumente à região selar. Localização. Tumores originados nas meninges são mais comuns que no parênquima nervoso ou no plexo coróide. A maioria dos tumores mesenquimais são supratentoriais, com exceção dos rabdomiossarcomas, que são mais freqüentemente infratentoriais. Condrossarcomas originam-se na base do crânio. Lipomas ocorrem tipicamente na linha média, como na região anterior do corpo caloso, placa quadrigeminal, região supraselar e hipotalâmica, e ainda no ângulo ponto-cerebelar e canal auditivo. Na medula envolvem mais o cone medular e filo terminal. Só lipomas são característicos na RM, devido ao forte hipersinal em T1 associado aos ácidos graxos. Macro. Sarcomas meníngeos são de textura firme e, apesar da aparente boa delimitação, tendem a invadir o cérebro. A superfície de corte é geralmente firme e carnosa, mas lesões de alto grau podem mostrar necrose e hemorragia. Micro. É superponível à dos tumores de mesma linhagem em outros sítios. Histogênese. Admite-se que os tumores mesenquimais afetando o SNC se originem das meninges e vasos crânio-espinais, ou das estruturas ósseas adjacentes. Tumores osteo-cartilaginosos ou miogênicos poderiam se originar: a) de heterotopias meníngeas destes tecidos (raras). b) células mesenquimais multipotenciais; c) aquisição de linhas adicionais de diferenciação em tumores fibrosos ou fibrohistiocíticos; d) a partir de teratomas. Prognóstico. A maioria dos tumores mesenquimais benignos podem ser totalmente ressecáveis e curáveis. Porém, os sarcomas intracranianos são agressivos e o prognóstico é ruim. Tipicamente há recidivas múltiplas e semeadura das leptomeninges à distância. Os rabdomiossarcomas são quase uniformemente fatais em 2 anos, mesmo com rádio- e quimioterapia agressivas. Sarcomas intracranianos também podem dar metástases extracranianas. Mesmo assim, os sarcomas intracranianos primários são ainda menos agressivos que os glioblastomas. Em uma série de 18 casos, a sobrevida de 5 anos foi estimada em 28% para os tumores de alto grau e 83% para os de baixo grau. Fonte.
Paulus W, Scheithauer BW, Perry A. Mesenchymal, non-meningothelial
tumours. In WHO Classification of Tumours of the Central
Nervous System. 4th Ed. Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, editors. International Agency for Research on Cancer, Lyon, 2007.
pp 173-7.
Constituem menos de 1% dos tumores intracranianos. Podem ocorrer em qualquer idade ou localização, espontaneamente ou secundariamente a radioterapia (estes mais comumente na região selar). Quando o envolvimento de tecidos é extenso (osso, dura, leptomeninges e cérebro) fica difícil definir a origem precisa. Os fibrossarcomas espontâneos formam um grupo heterogêneo quanto à idade, localização e aspectos macro- e microscópico. Alguns são massas aderidas à dura com tendência à infiltração óssea. Outros são lesões leptomeníngeas difusas. Outros formam uma massa intracerebral bem definida, abaixo da superfície cortical. Microscopicamente, alguns parecem-se com fibrossarcomas de outras localizações, com feixes entrelaçados de células fusiformes, e produção de colágeno e reticulina. Outros tumores têm células mais pleomórficas, menos alongadas, associadas a pouco estroma, e lembram mais o fibrohistiocitoma maligno. Só raros sarcomas meníngeos primários originam-se de músculo liso (leiomiossarcomas). Imunohistoquímica. Sarcomas expressam vimentina. São negativos para GFAP, proteína S-100 e EMA. Microscopia eletrônica. Os fibroblastos neoplásicos são variáveis em configuração, mas há tipicamente células bipolares. Cisternas dilatadas de retículo endoplasmático rugoso são conspícuas. Geralmente, as células contêm poucos filamentos intermediários e não têm junções intercelulares especializadas, nem membrana basal. Prognóstico. Fibrossarcomas tendem a apresentar disseminação liquórica e podem dar metástases sistêmicas. Em um estudo de 9 pacientes, a sobrevida mediana foi 7,5 meses. Fonte.
Burger, PC, Scheithauer BW, Vogel FS. Surgical Pathology of the Nervous
System and its Coverings. 4th Ed. Churchill Livingstone, New York,
2002. pp 83-4; 315-6.
Mixofibrossarcoma (ou fibrohistiocitoma maligno mixóide). Mixofibrossarcoma é uma entidade distinta e bem definida que mostra amplo espectro de grau histológico. Tumores na extremidade menos diferenciada ou de alto grau do espectro assemelham-se ao assim chamado fibrohistiocitoma maligno pleomórfico, em termos de celularidade, atipias e pleomorfismo. Esta foi a origem do termo fibrohistiocitoma maligno mixóide. Clinicamente, o mixofibrossarcoma afeta principalmente adultos da 6ª. à 8ª. décadas, embora a variação de idade seja ampla, com discreto predomínio no sexo masculino. A grande maioria destes tumores origina-se em membros. Origem retroperitonial é extremamente infreqüente. Quando este padrão morfológico é encontrado em tumores intraabdominais, cabe o diagnóstico diferencial com o lipossarcoma pouco diferenciado. Cerca de 2/3 dos casos origina-se em tecidos subcutâneos, não em regiões profundas. Há tendência a crescimento multinodular e difusamente infiltrativo, o que leva freqüentemente a uma subestimativa do real tamanho da neoplasia. A sobrevida de 5 anos está na faixa de 60-70%, mas há correlação entre a sobrevida e o grau histológico. As lesões de grau mais baixo não têm capacidade para metastatizar, mas podem progredir a graus mais altos. Lesões de grau mais alto têm tendência a metástases linfonodais, além das pulmonares e ósseas. Histologicamente, tumores de todos os graus mostram áreas hipocelulares, contendo vasos curvilíneos de paredes finas. Nestas áreas, são encontradas células atípicas pequenas, fusiformes ou estrelares, com núcleos hipercromáticos e citoplasma mal definido, por vezes vacuolado. Esses pequenos vacúolos bolhosos contêm mucina em vez de lípide e estas células são chamadas às vezes de pseudolipoblastos. A celularidade e o pleomorfismo vão em paralelo com o grau histológico. Para qualificar como mixofibrossarcoma, pelo menos 5 a 10% do tumor deve conter um proeminente estroma mixóide. O mucopolissacáride principal na matriz mixóide é ácido hialurônico. As células fusiformes ou estrelares têm características ultraestruturais de fibroblastos ou, ocasionalmente, miofibroblastos. Em imunohistoquímica, isto se correlaciona com a positividade focal para actina em alguns casos, principalmente lesões de alto grau com áreas mais sólidas. Há também positividade difusa e consistente para vimentina. Outros marcadores são geralmente negativos. Em lesões de alto grau, áreas do tumor são indistinguíveis do padrão inespecífico do fibrohistiocitoma maligno pleomórfico. Em ME, estas células bizarras podem aparecer indiferenciadas ou semelhantes a histiócitos. Fonte. Fletcher CDM. Soft tissue tumors. Chapter 24 In Fletcher CDM (editor). Diagnostic Histopathology of Tumors. 2nd Ed., Churchill Livingstone 2000, p. 1506. |
| Textos complementares: | Sarcomas cerebrais | Fibrossarcoma | Mixofibrossarcoma | Diferencial: gliossarcoma |
| Neuropatologia
- Graduação |
Neuropatologia -
Casos Complementares |
Neuroimagem
- Graduação |
Neuroimagem -
Casos Complementares |
Correlação
Neuropatologia - Neuroimagem |
| Índice alfabético - Neuro | Adições recentes | Banco de imagens - Neuro | Patologia - outros aparelhos | Pages in English |
|
|
|
|