| Tumor teratóide/rabdóide
atípico (ATRT).
Definição.
Tumor altamente maligno constituído pelo menos em parte por células
grandes ou rabdóides, com imunofenótipos diversos representando
os três folhetos embrionários, habitualmente linhagens neuroectodérmica,
epitelial e mesenquimal. São grau histológico IV da
OMS.
Idade, localização.
Correspondem a 1-2% dos tumores cerebrais pediátricos e cerca de
10% dos tumores cerebrais em crianças pequenas. Apresentam-se
geralmente nos dois primeiros anos de vida, com predomínio no sexo
masculino 3 : 2. São raros em adultos. Ocorrem no compartimento
intracraniano, com predomínio na fossa posterior (2/3). Contudo,
em duas séries, os supratentoriais superaram os infratentoriais
(4/3). Os supratentoriais situam-se nos hemisférios cerebrais, menos
freqüentemente nos ventrículos, região supraselar e
região pineal. A maioria dos casos em fossa posterior são
intracerebelares, mas podem ter seu epicentro no ângulo ponto-cerebelar
ou no tronco cerebral. Exemplos espinais são raros. Há
relatos de coexistência de tumores rabdóides renais e tumores
intracranianos. Estes podem ser de pequenas células como meduloblastomas
ou pineoblastomas, mas também tumores rabdóides.
Clínica.
Os sintomas de apresentação em crianças pequenas são
letargia, vômitos, emagrecimento. Cefaléia e hemiparesia predominam
em crianças mais velhas.
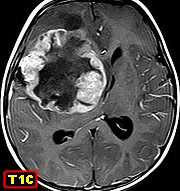
Imagem.
As lesões são volumosas, às vezes císticas
e/ou hemorrágicas, dão hipersinal no TR longo e captam contraste
no componente sólido. São mais comumente necróticas
que os meduloblastomas, que são o principal diagnóstico diferencial.
Cerca de ¼ já têm disseminação liquórica
quando do diagnóstico.
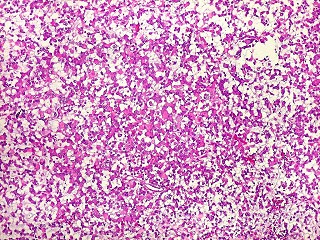
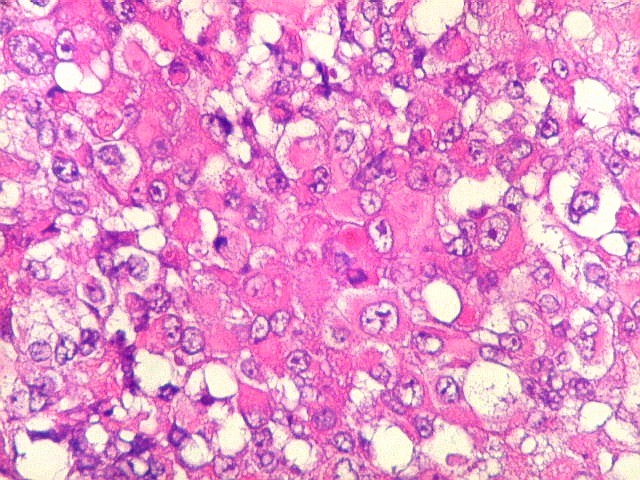
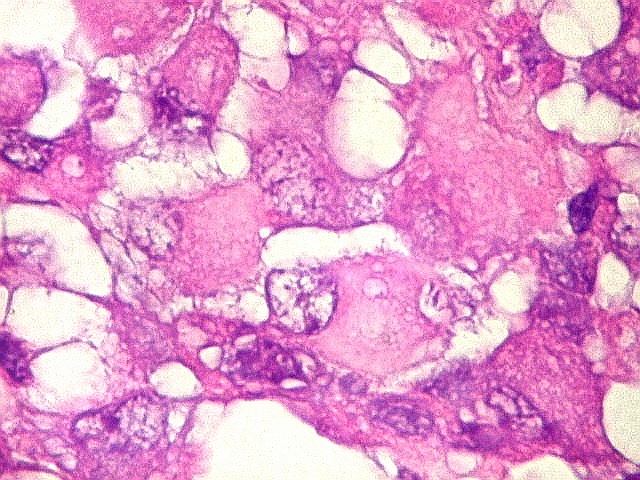
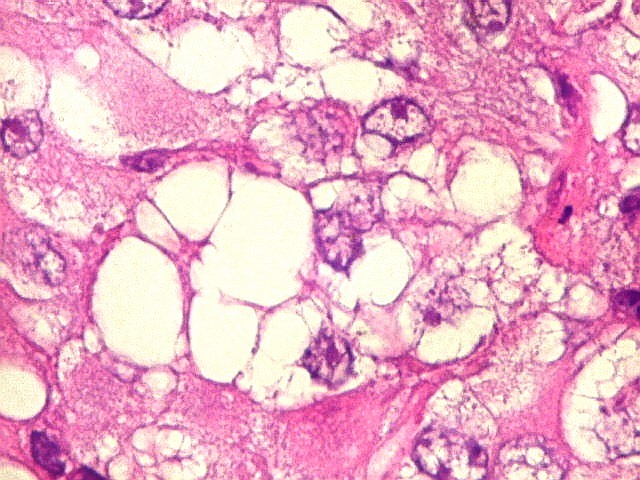
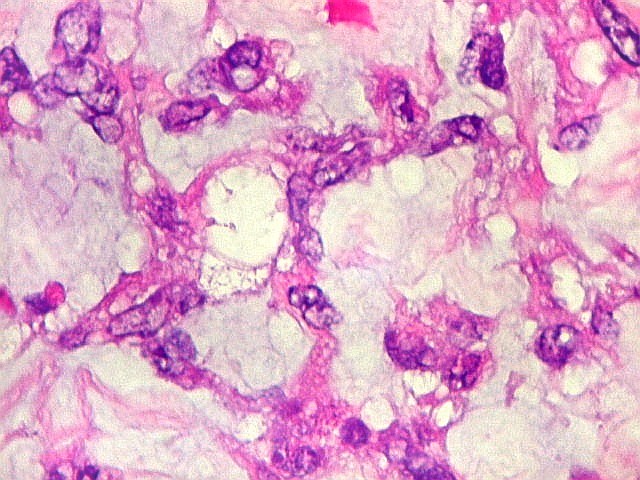
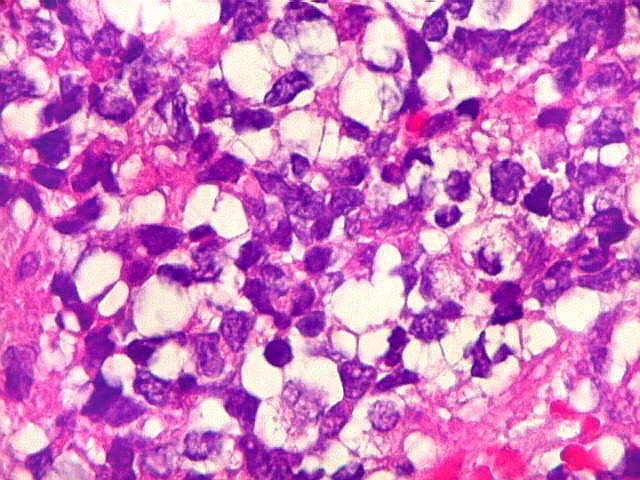
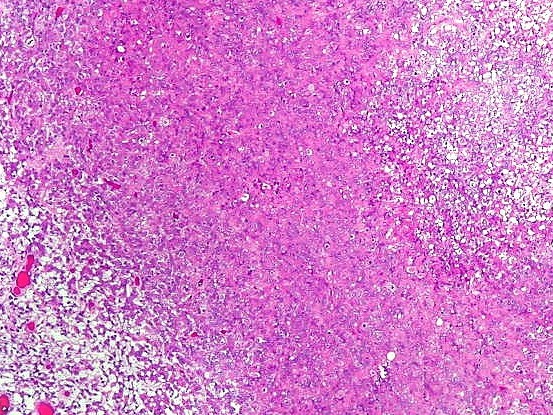
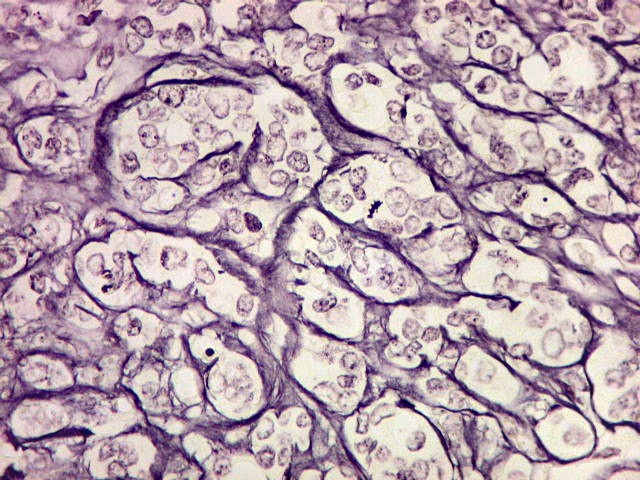
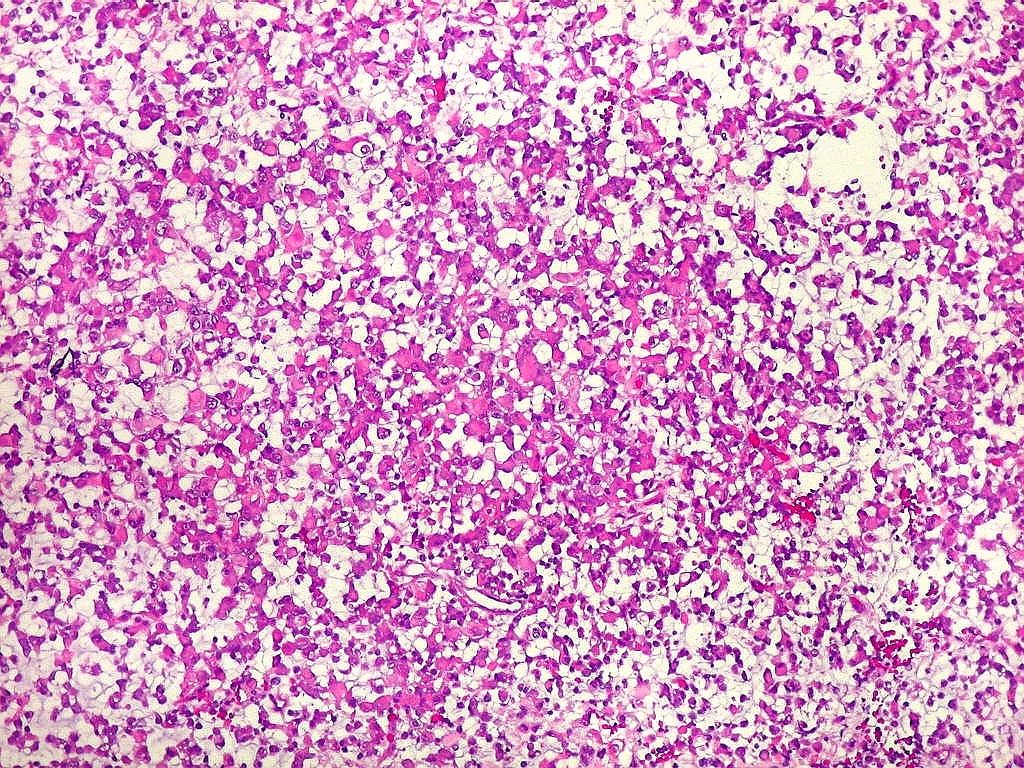
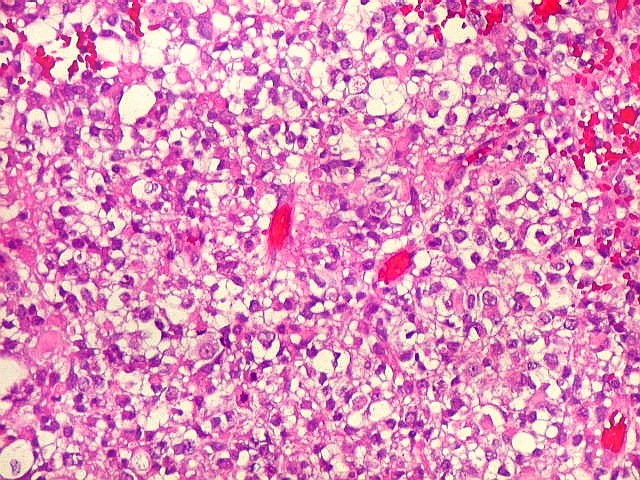
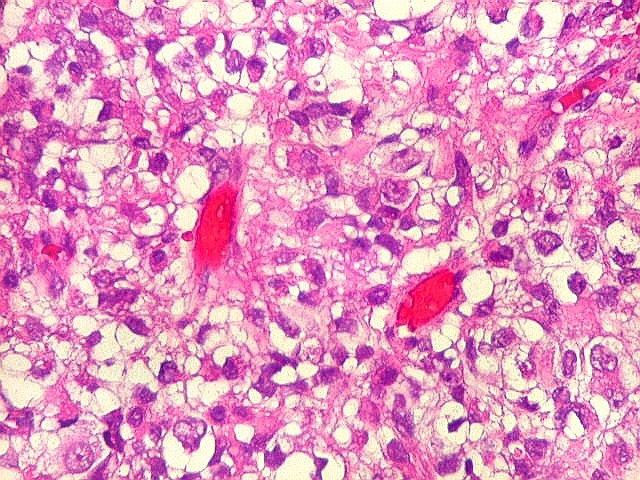
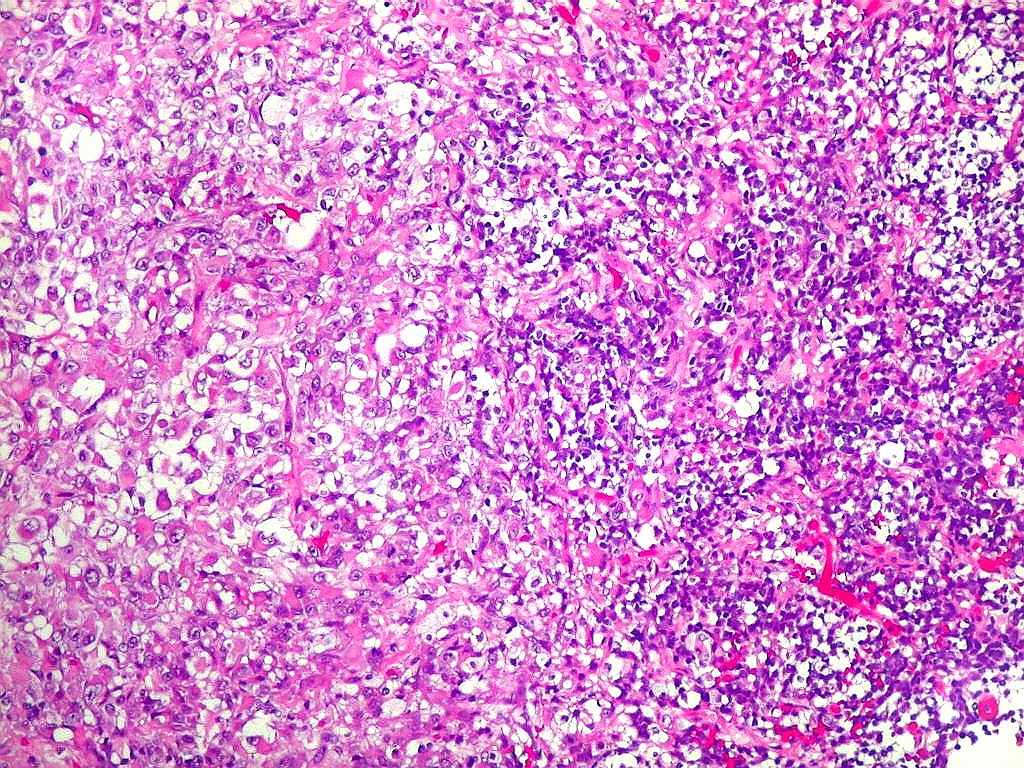
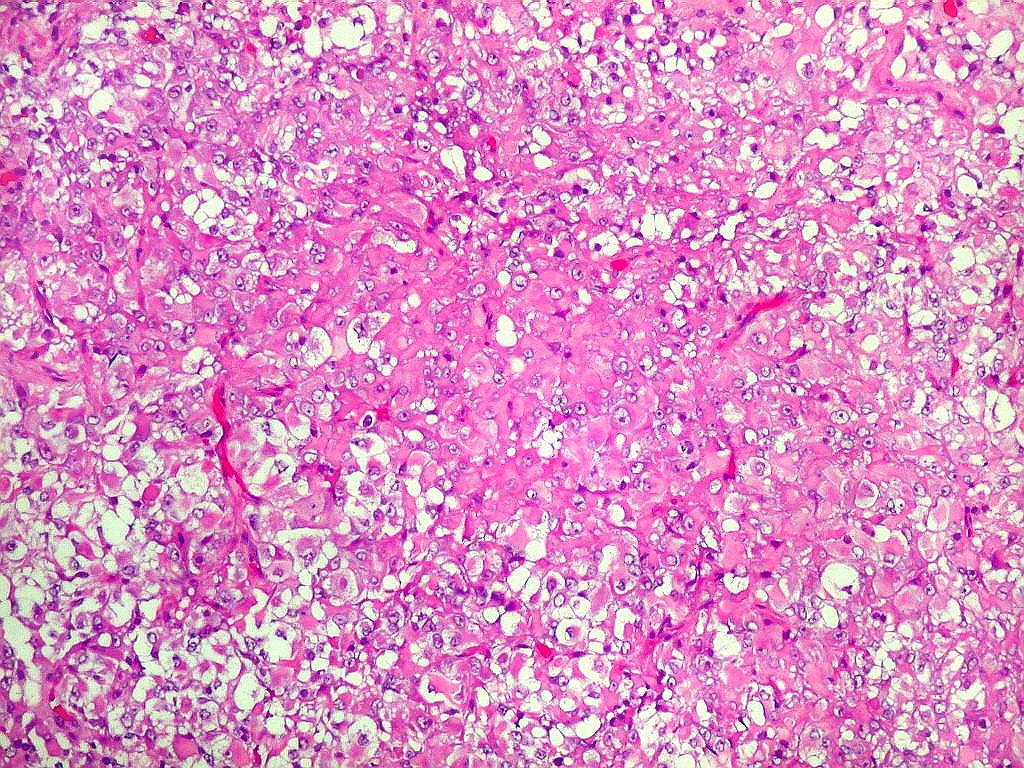
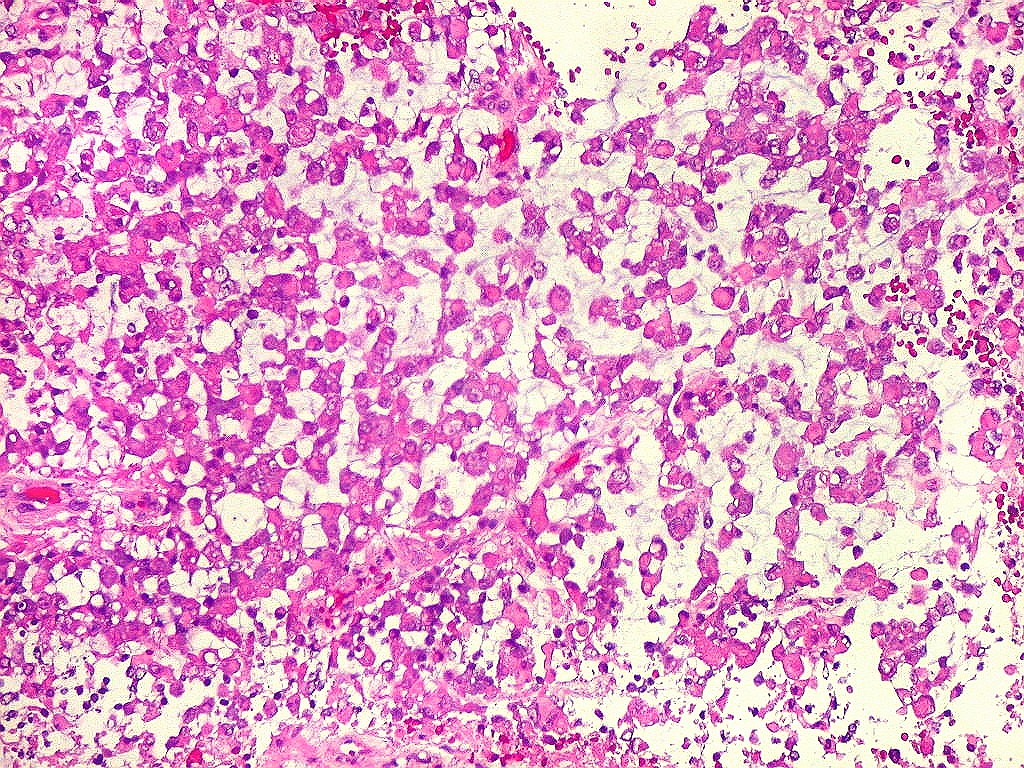
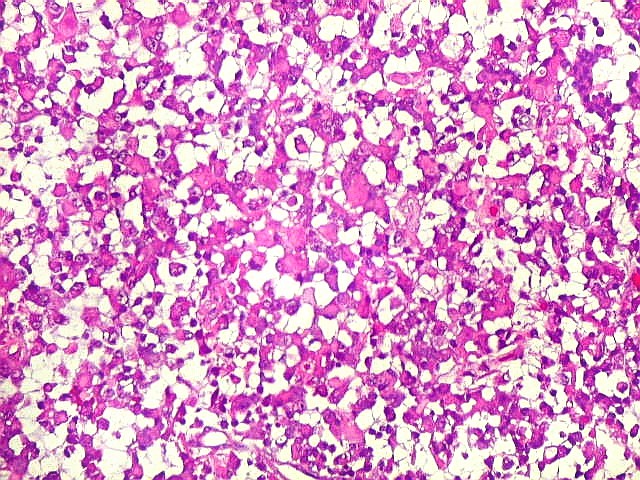
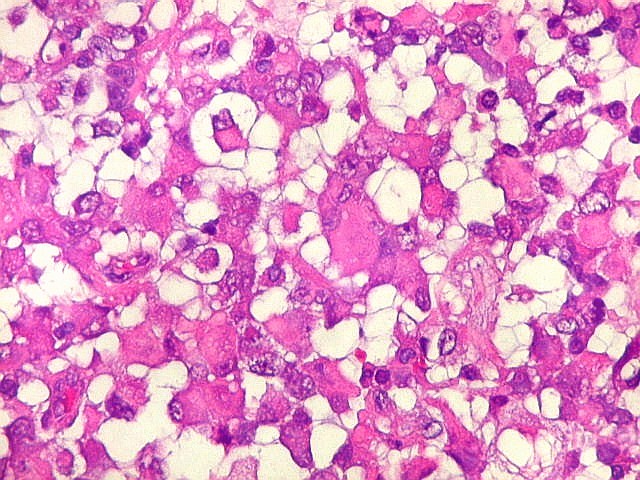
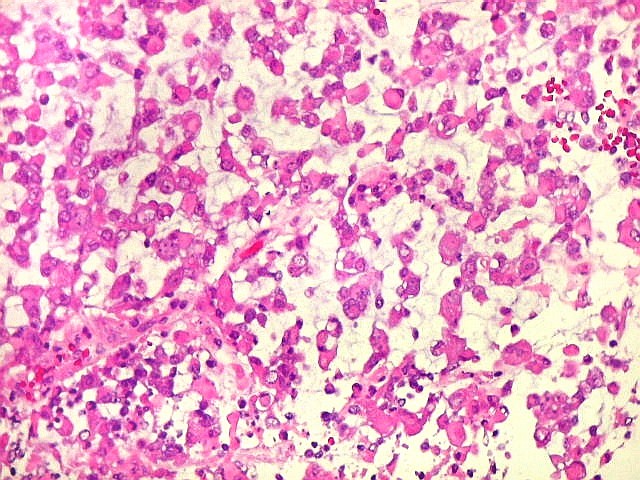
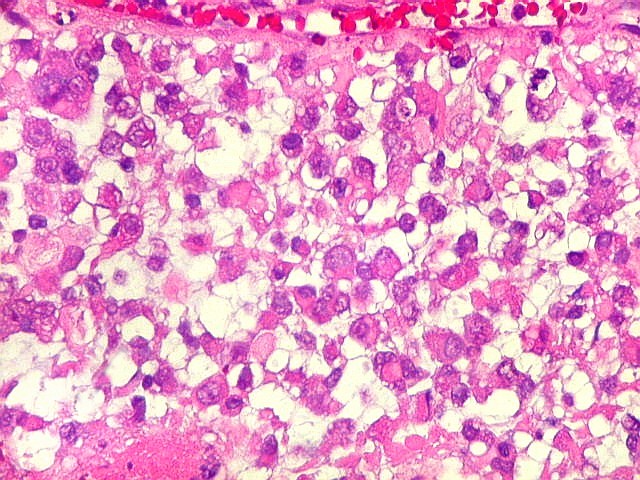
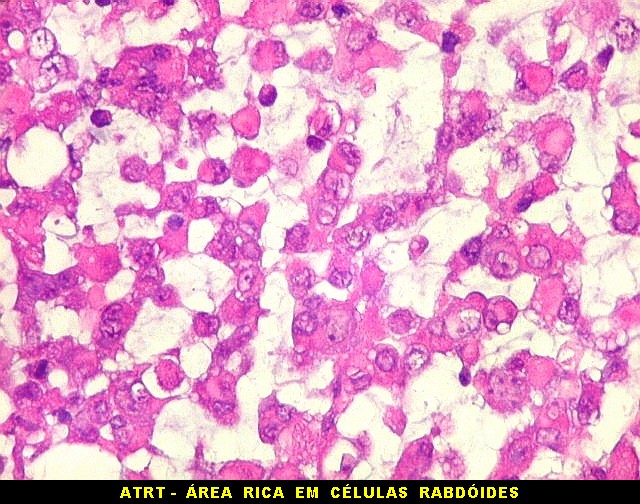
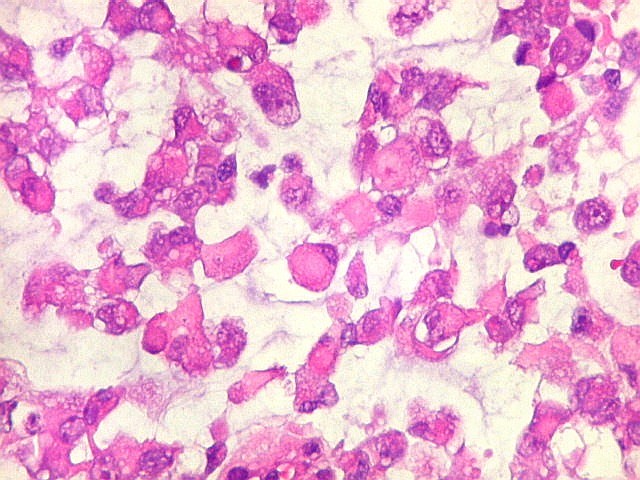
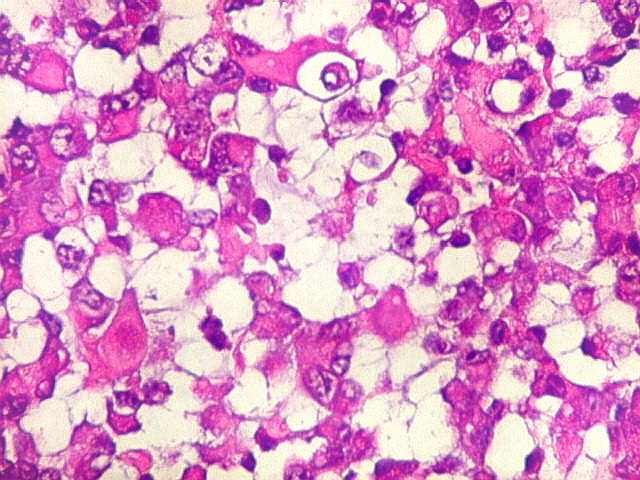
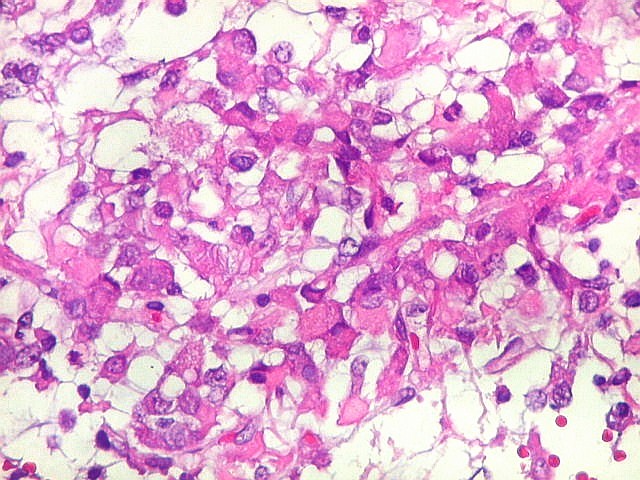
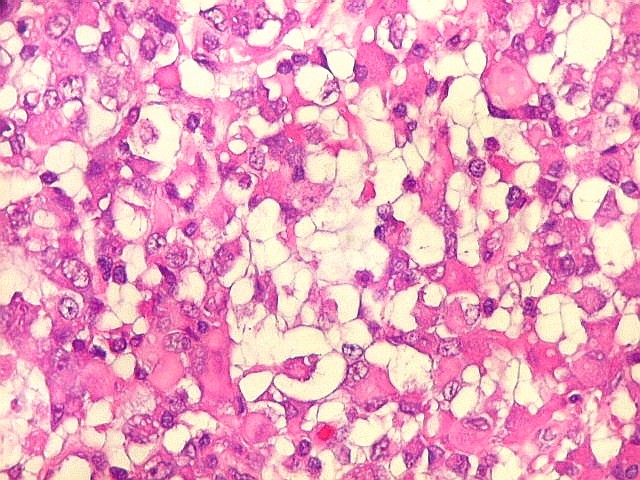
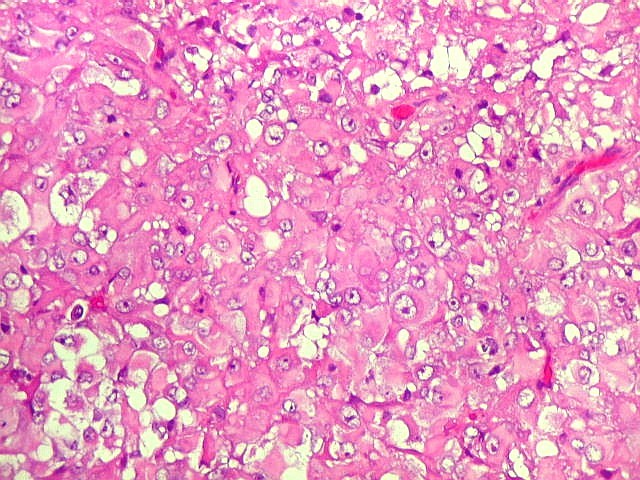
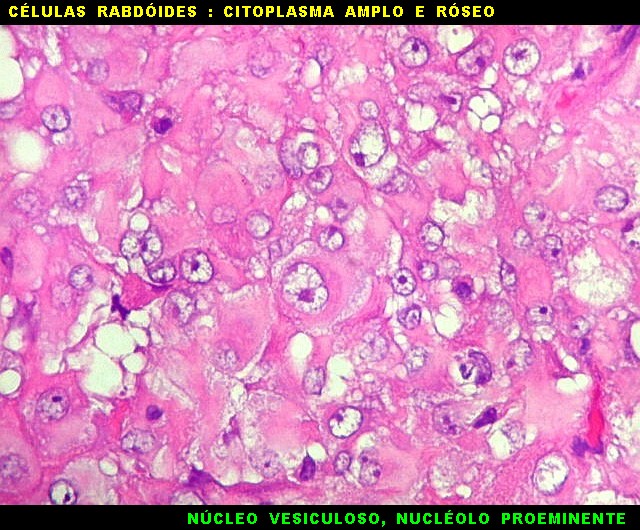
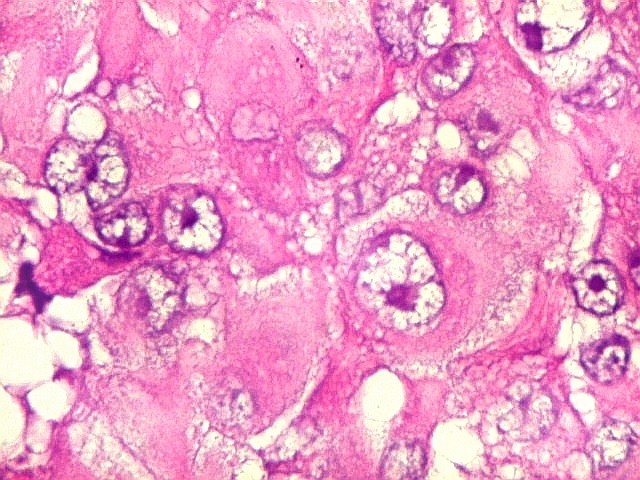
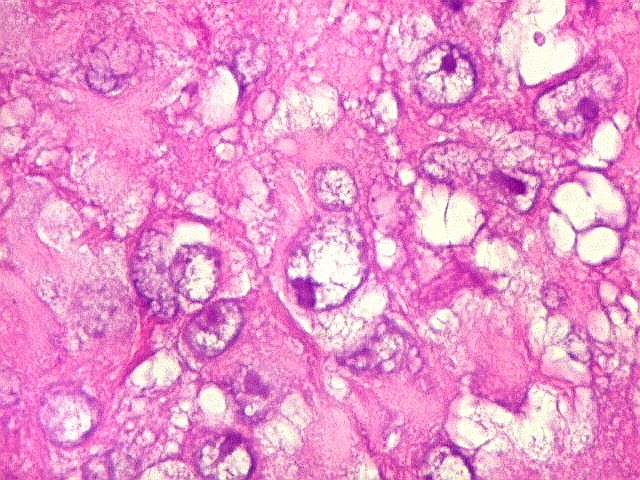
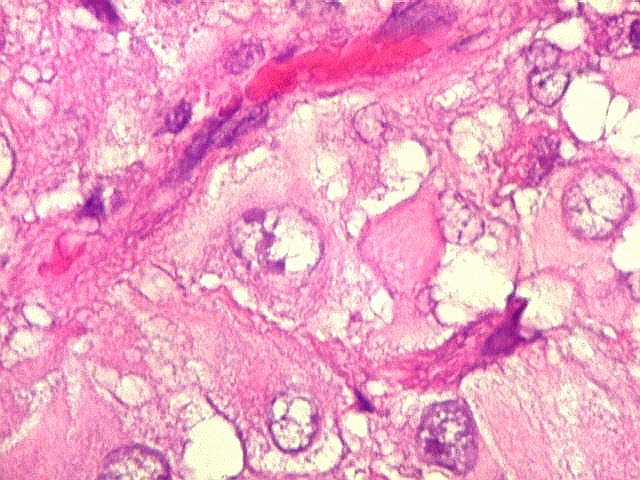
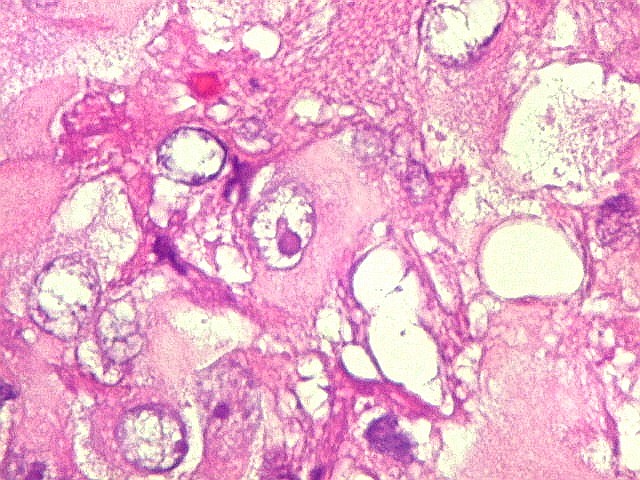
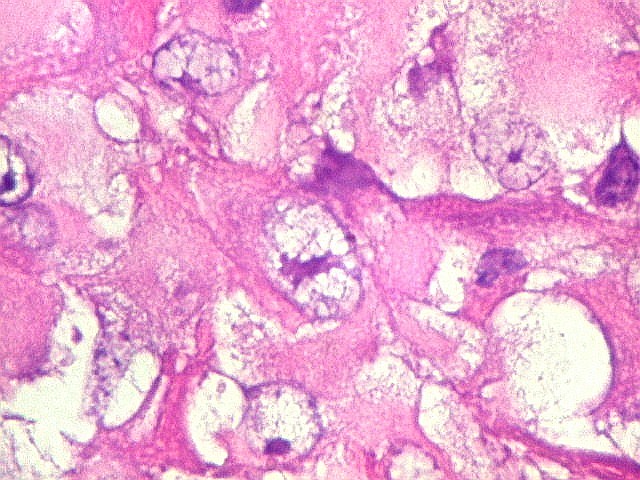
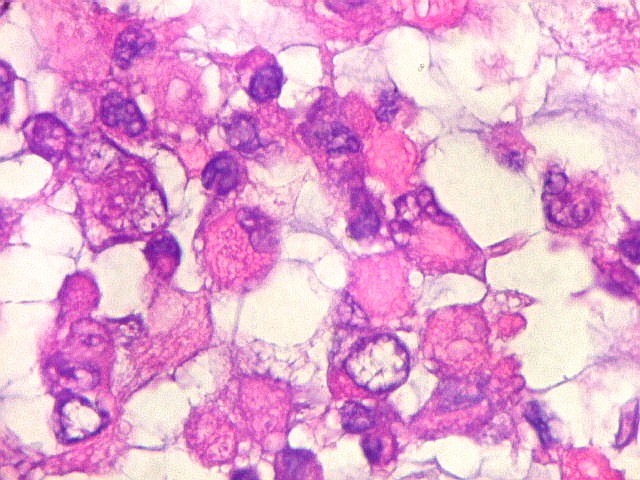
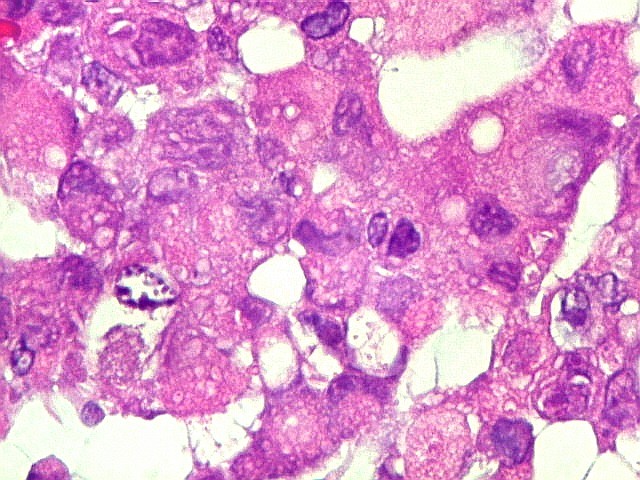
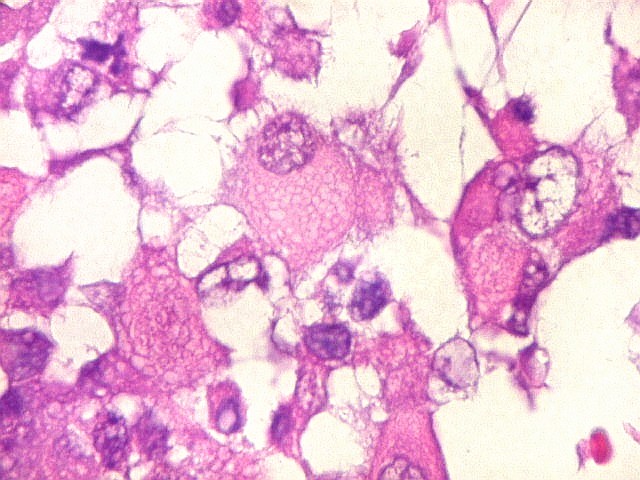
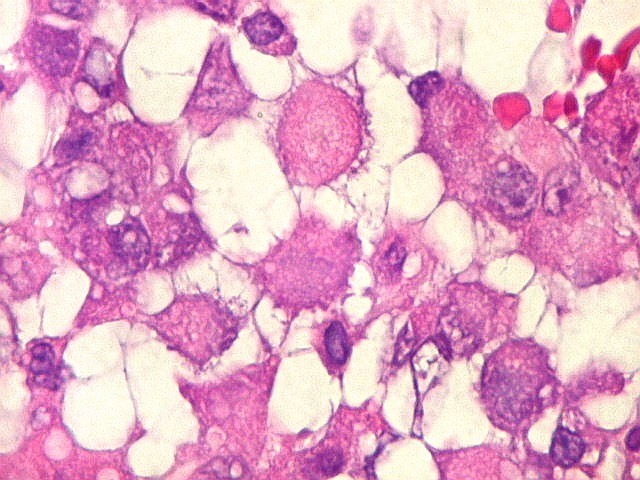
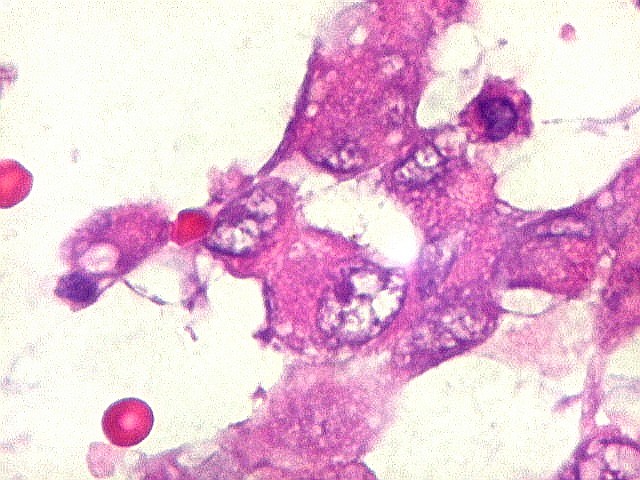
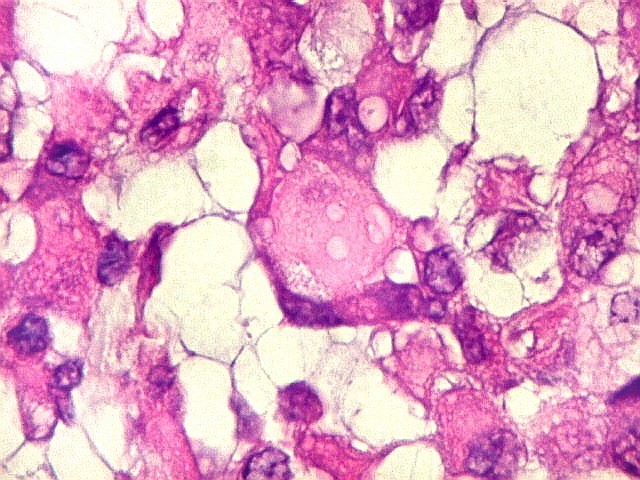
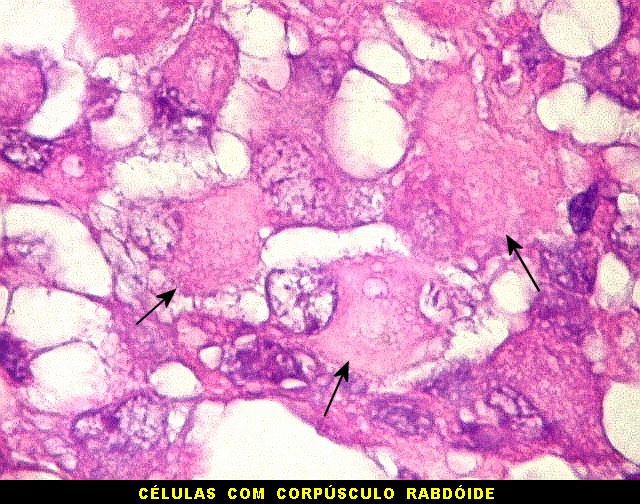
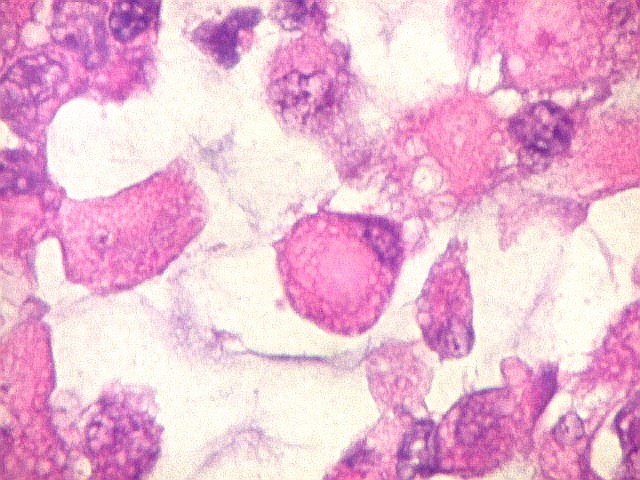
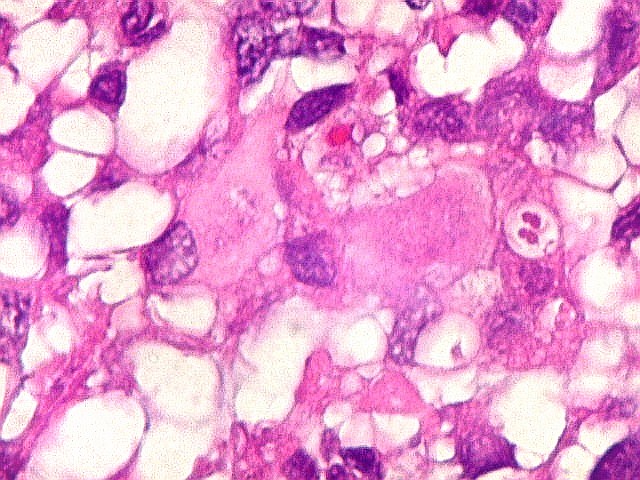
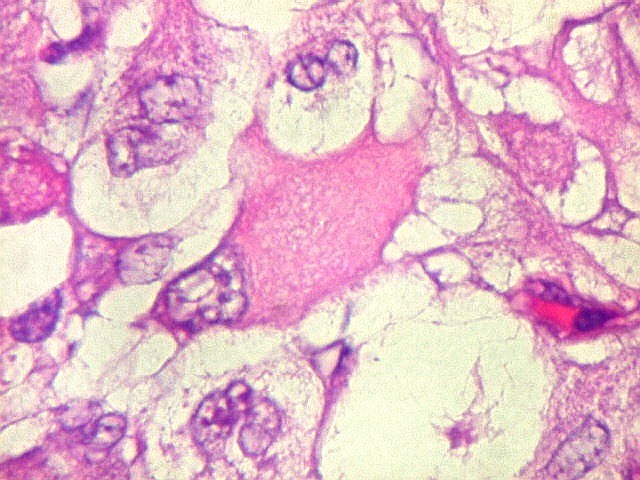
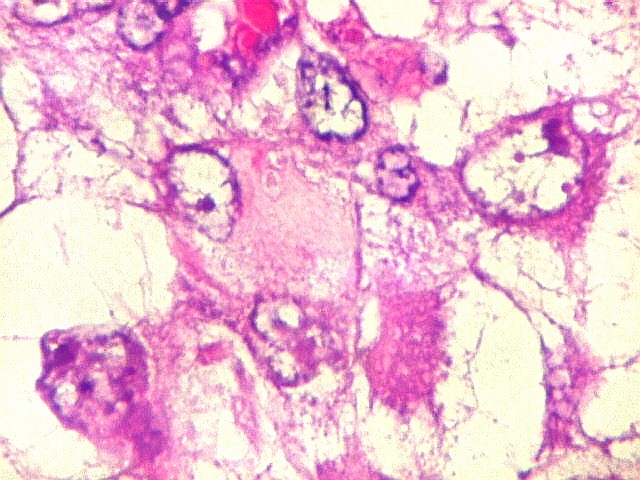
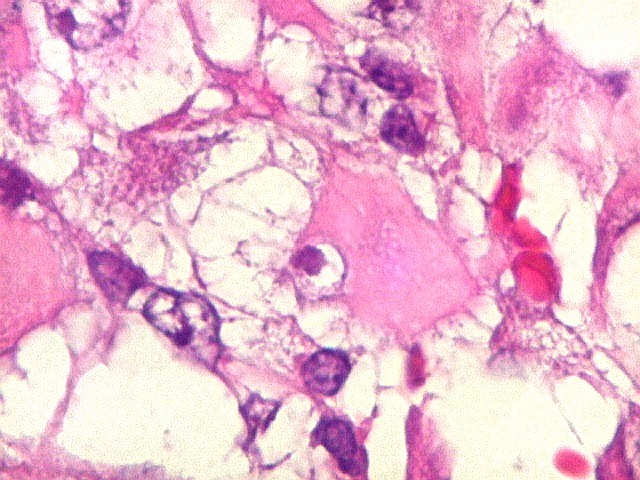
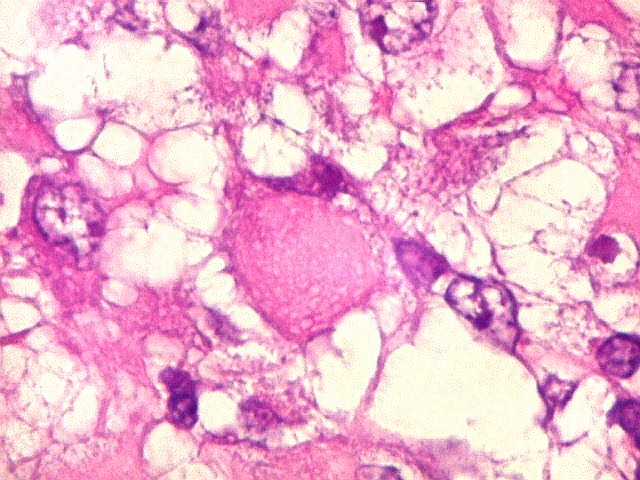
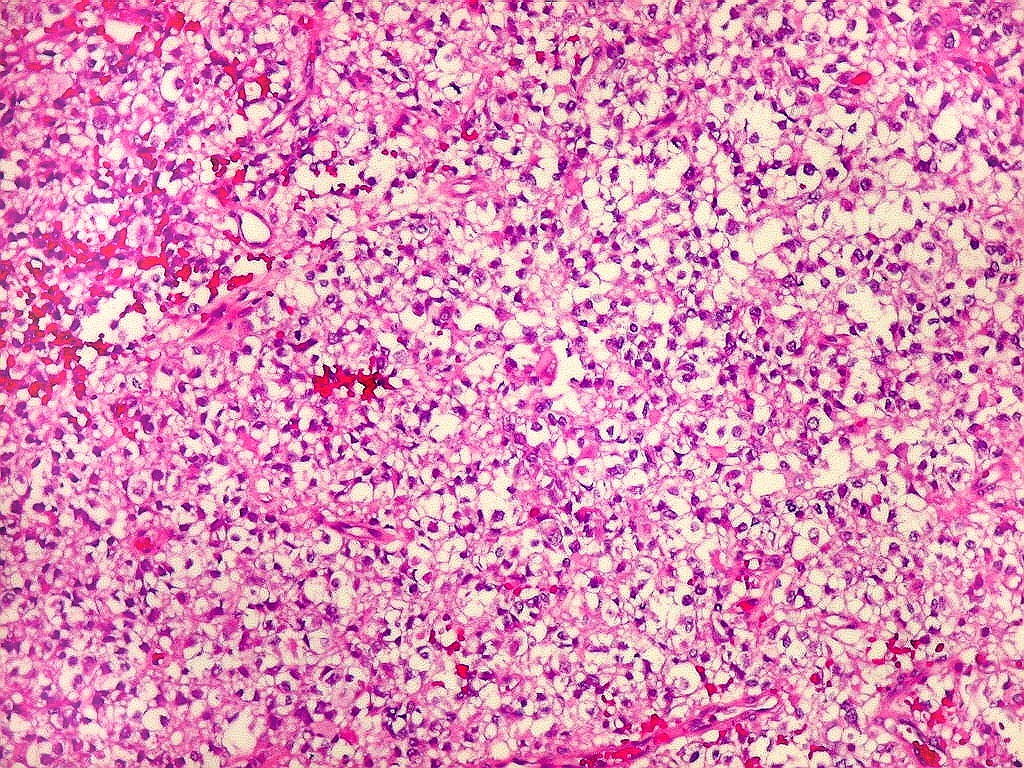
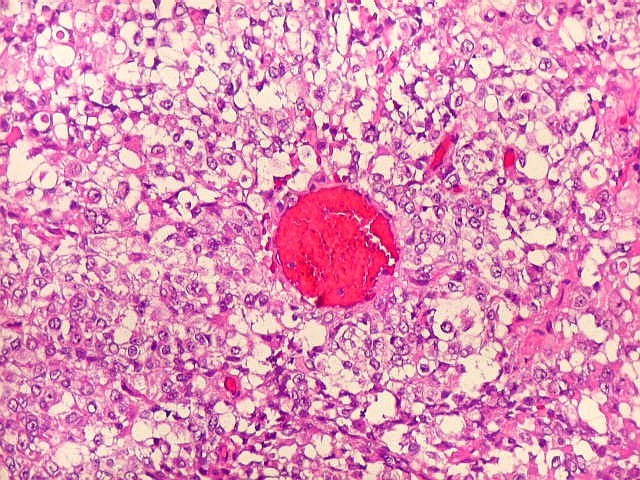
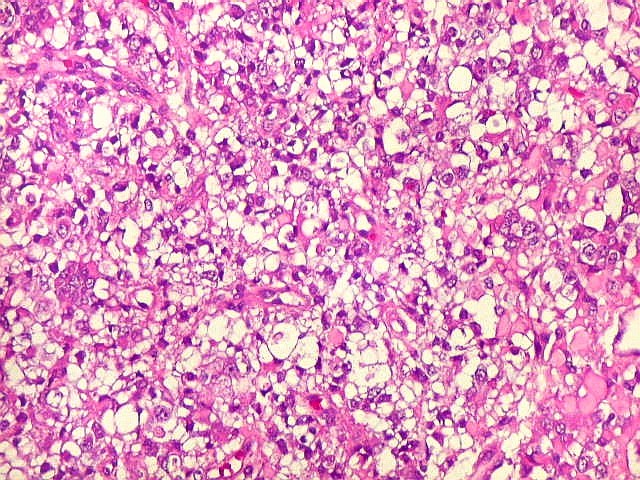
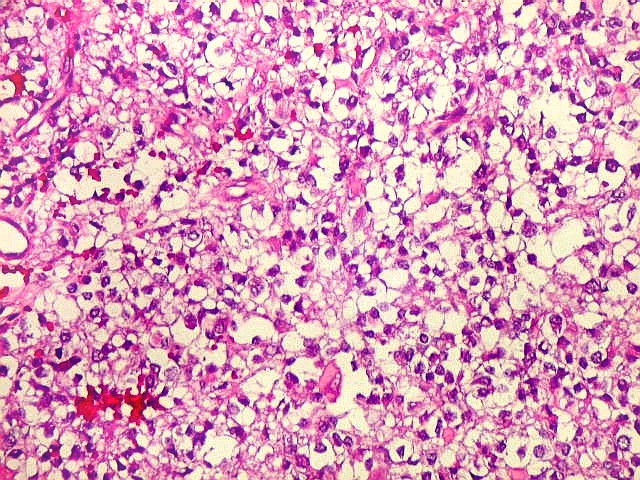
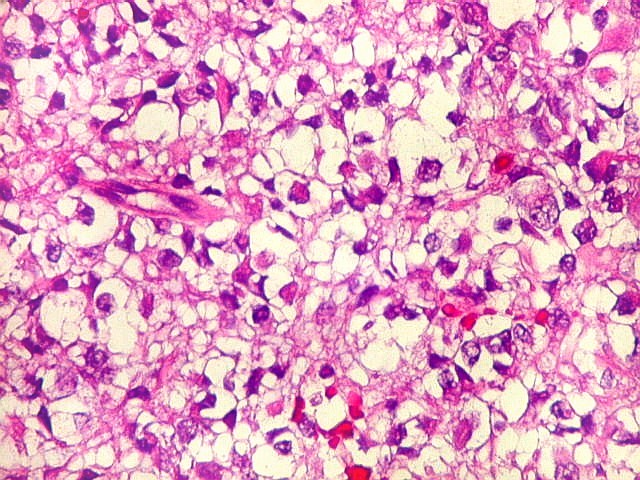
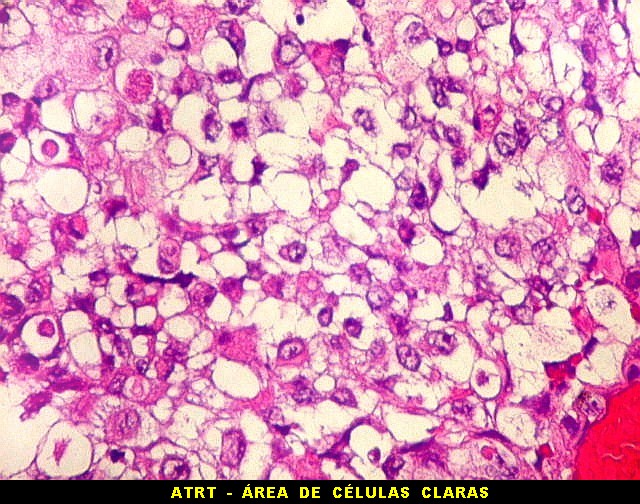
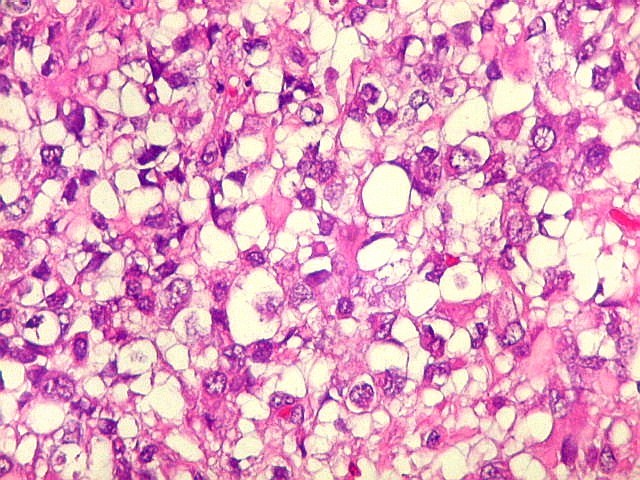
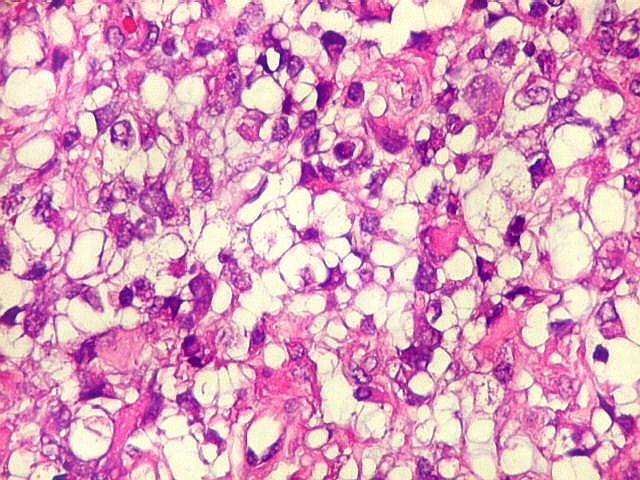
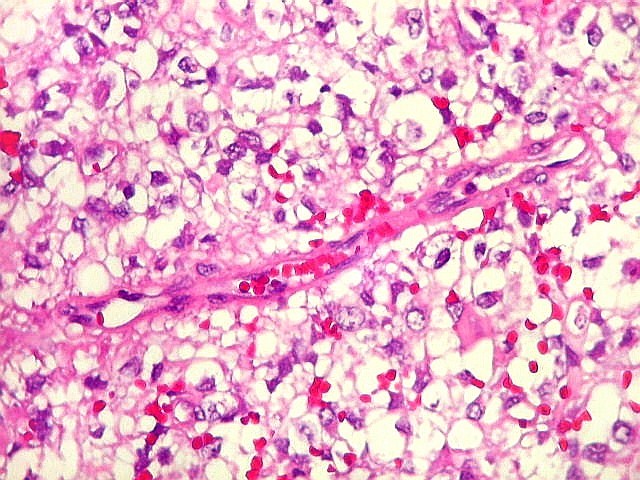
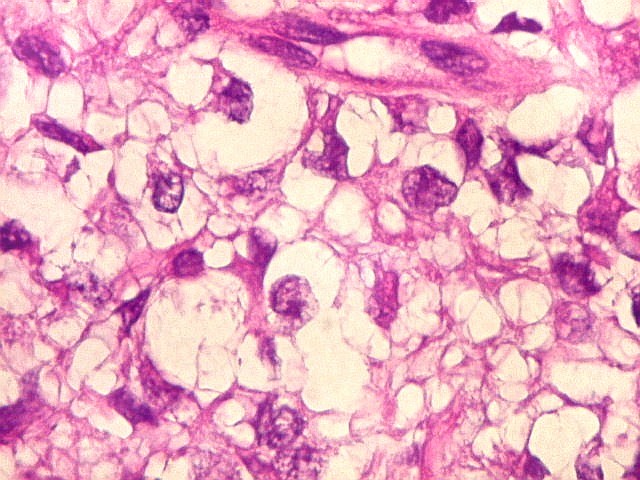
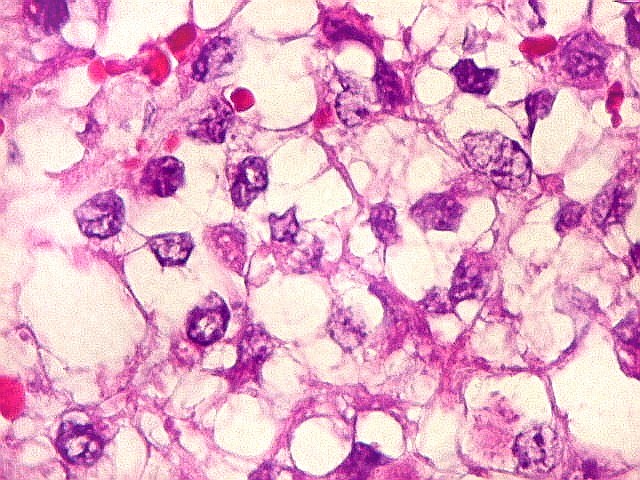
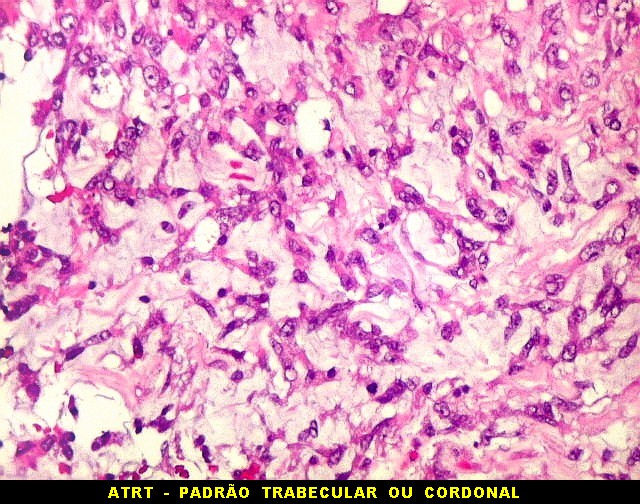
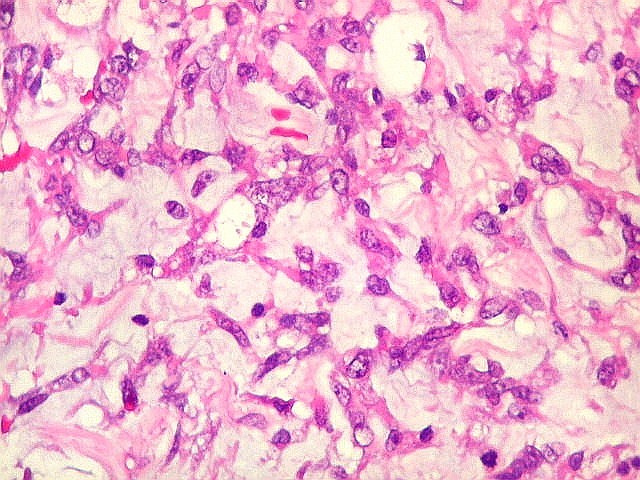
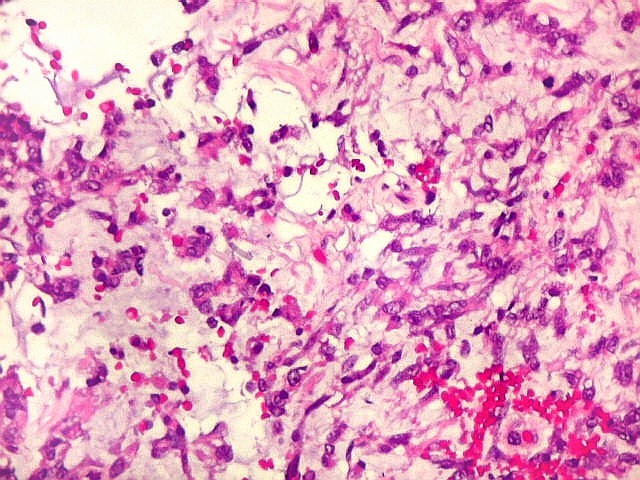
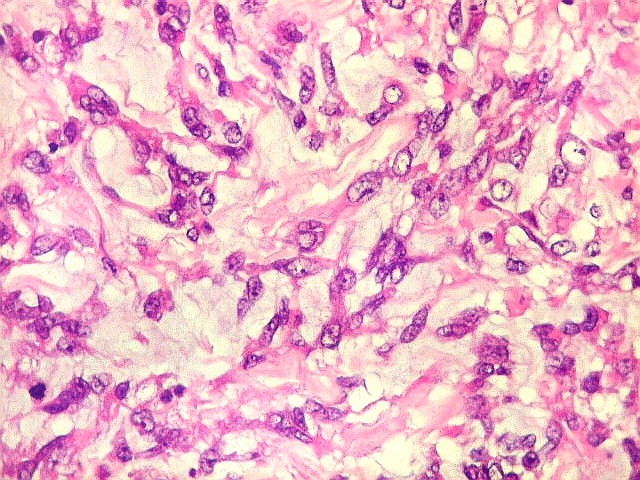
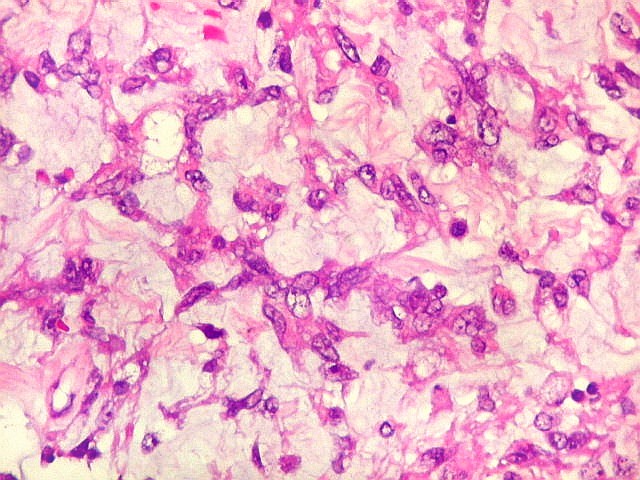
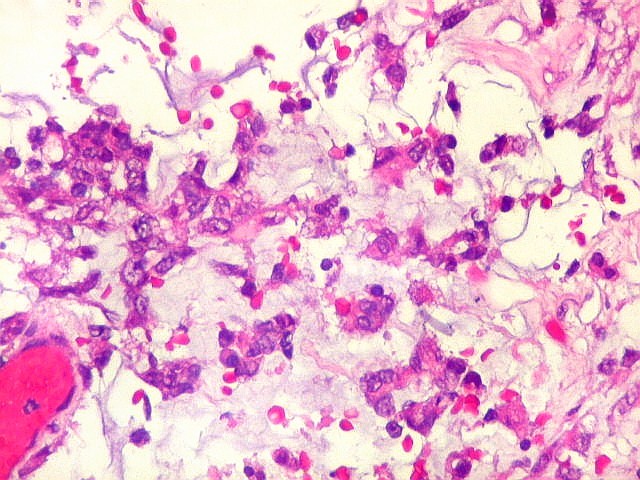
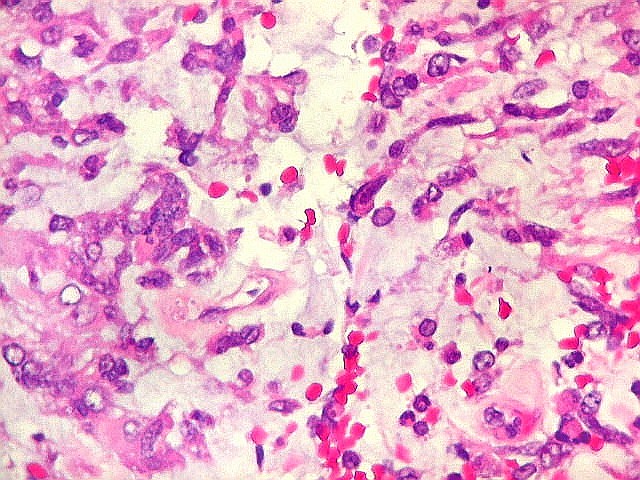
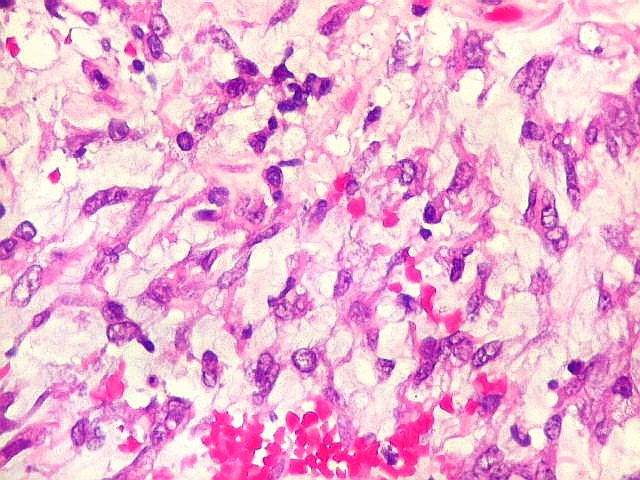
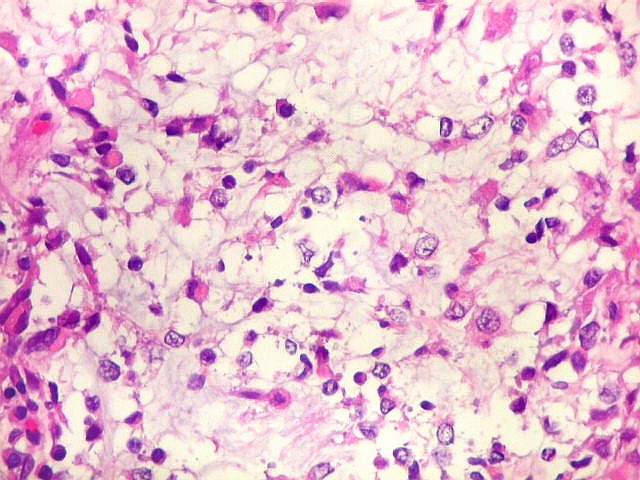
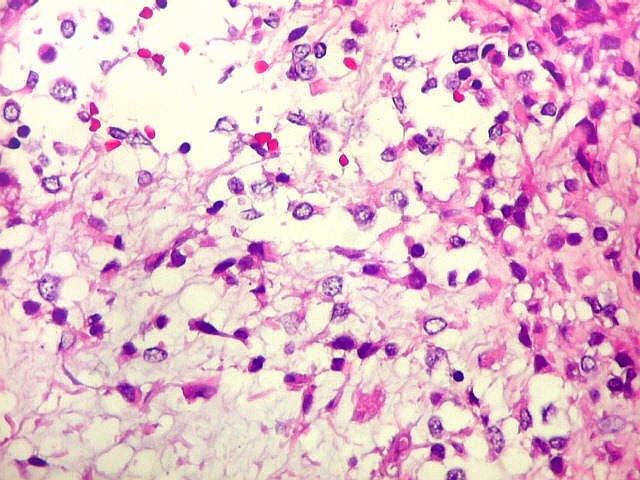
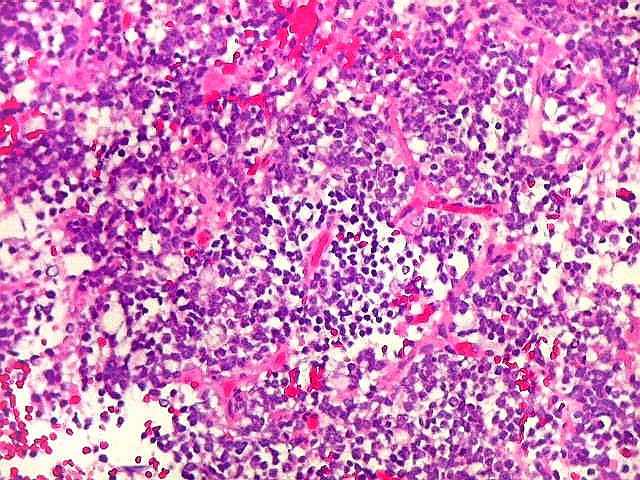
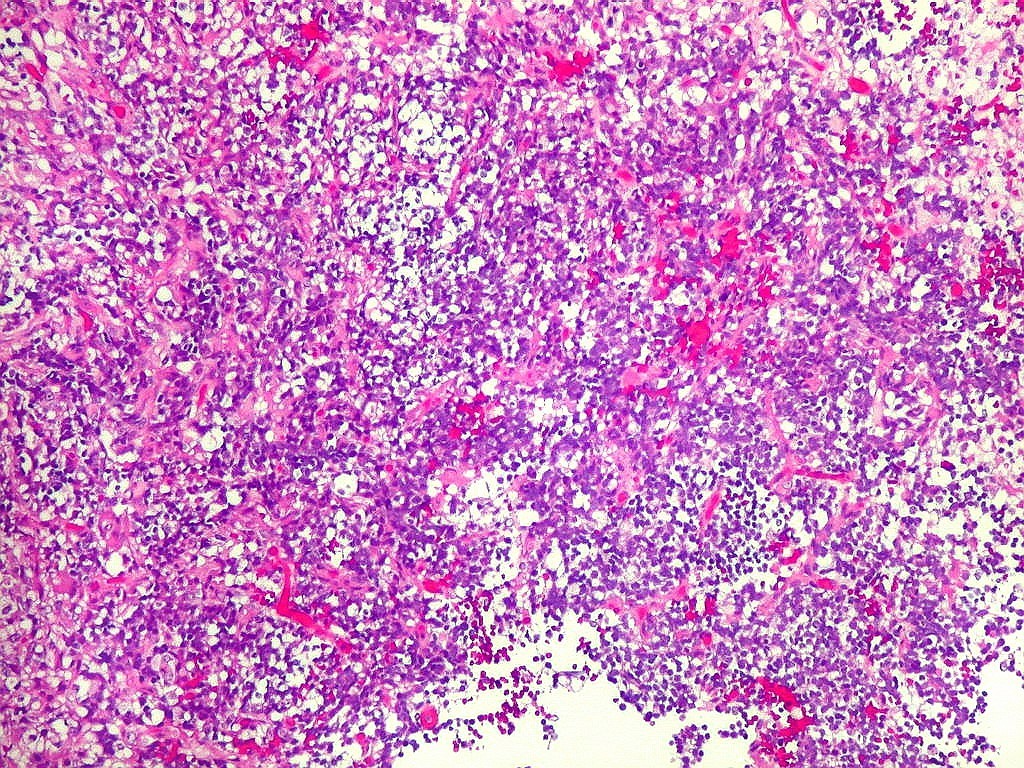
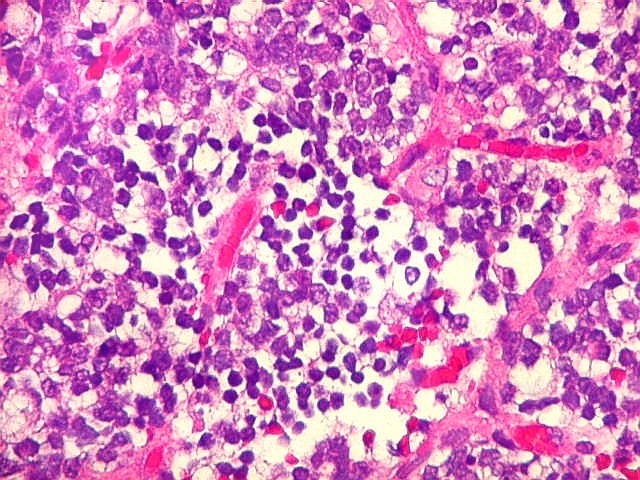
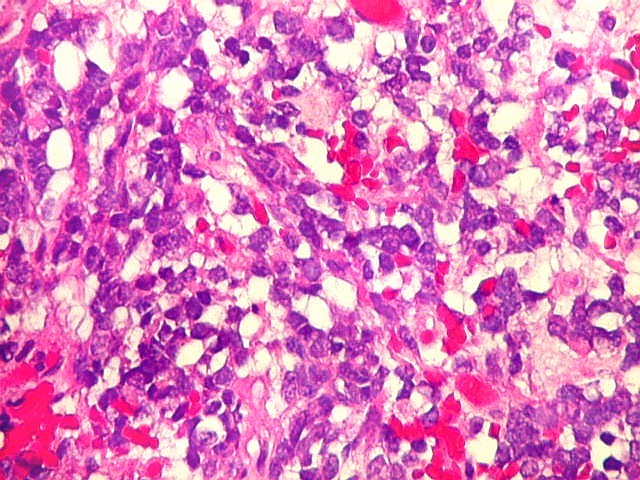
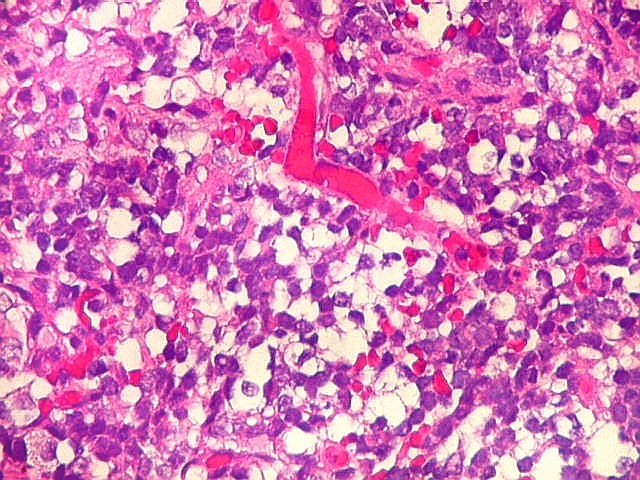
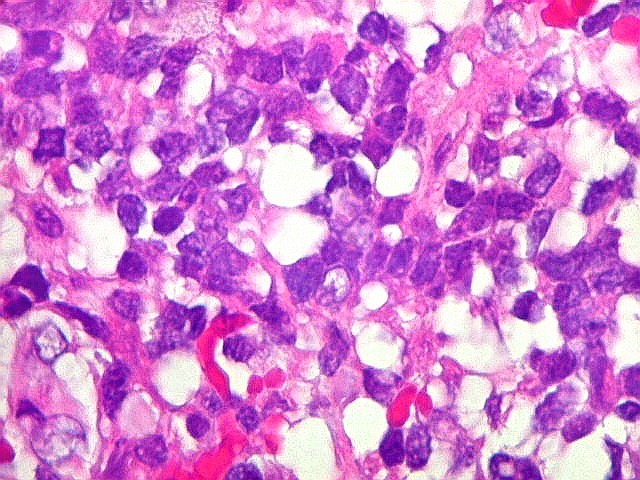
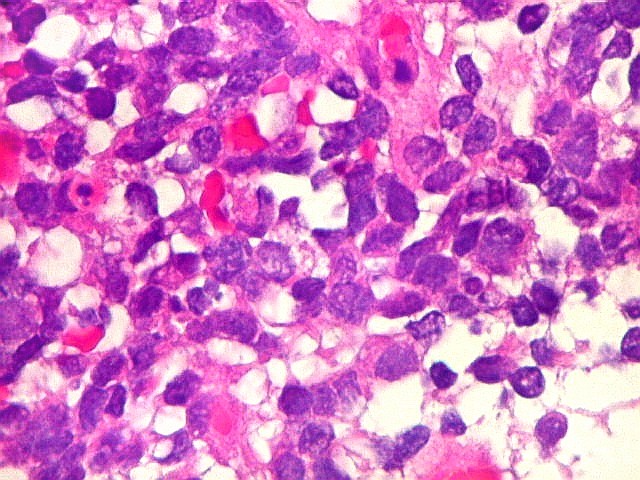
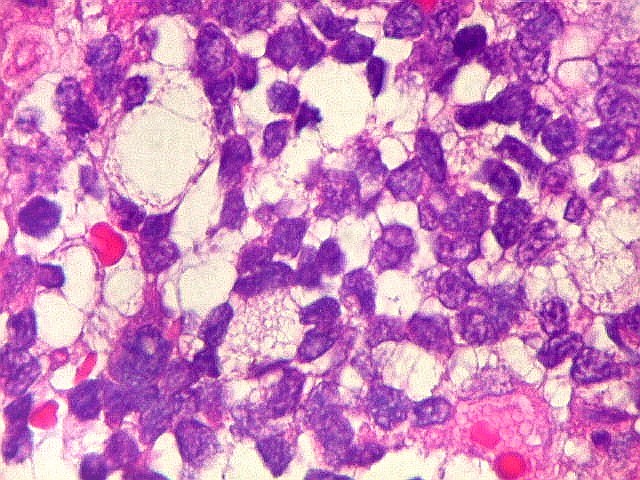
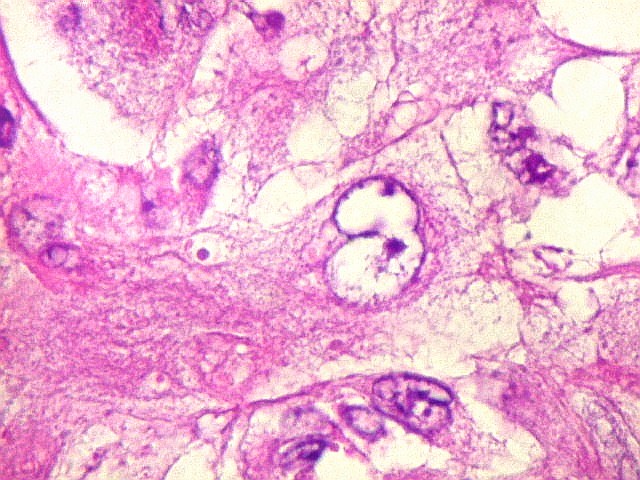
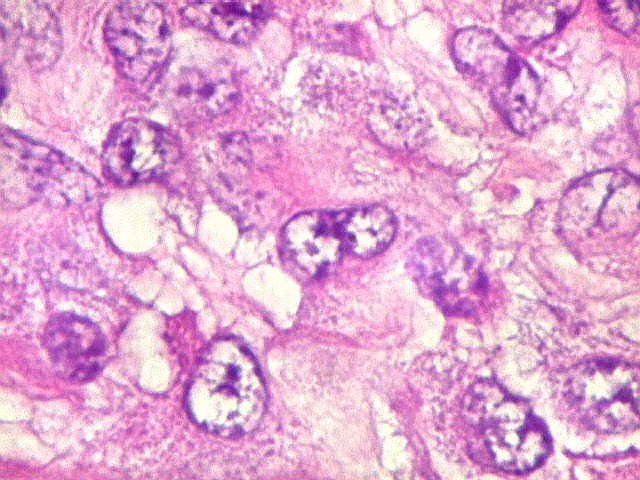
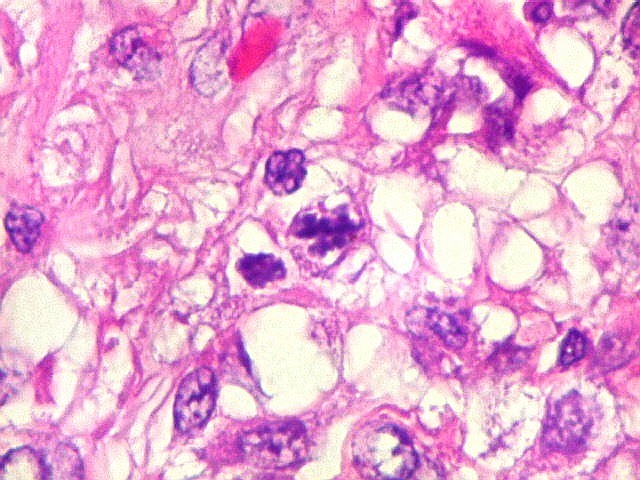
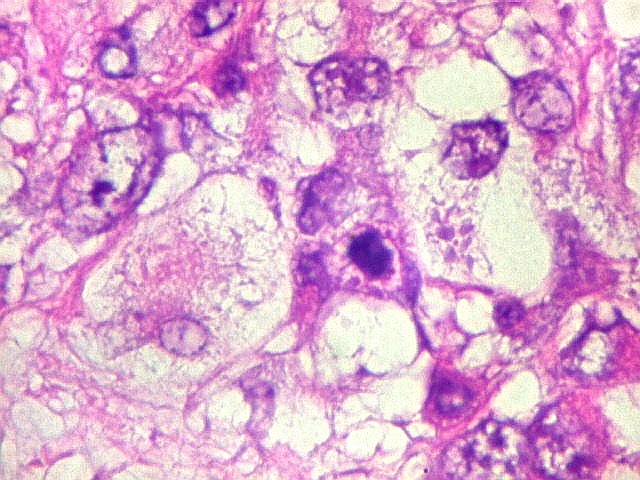
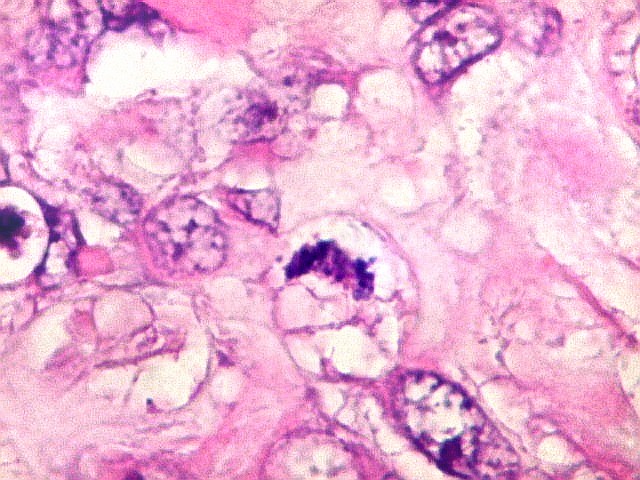
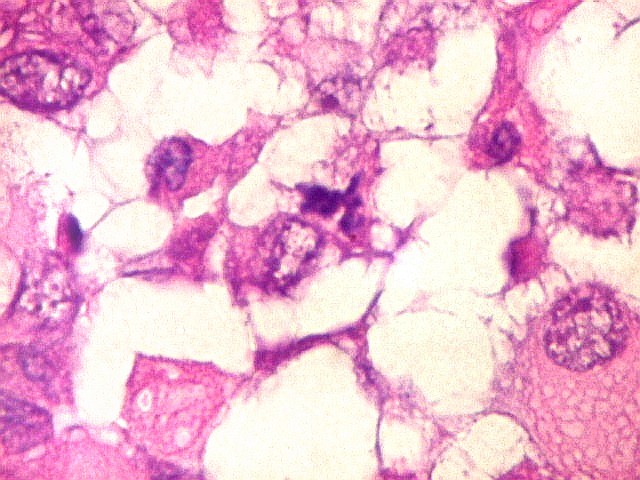
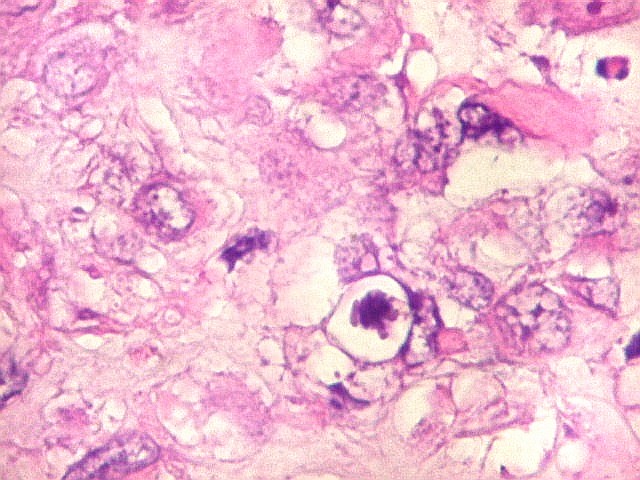
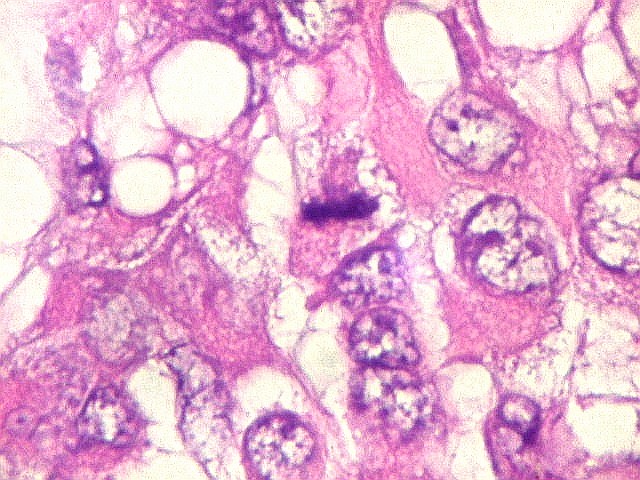
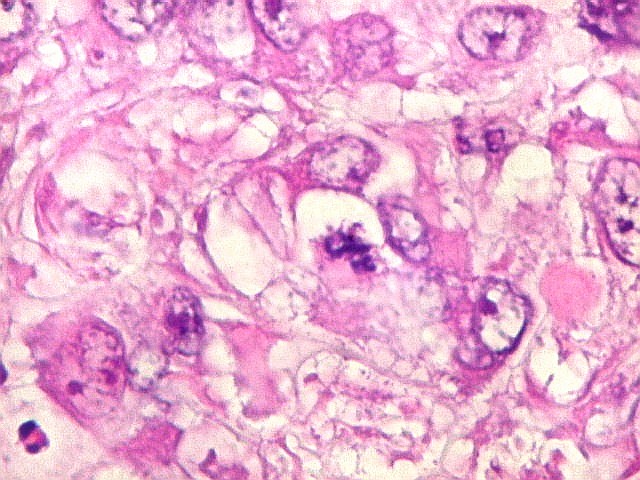
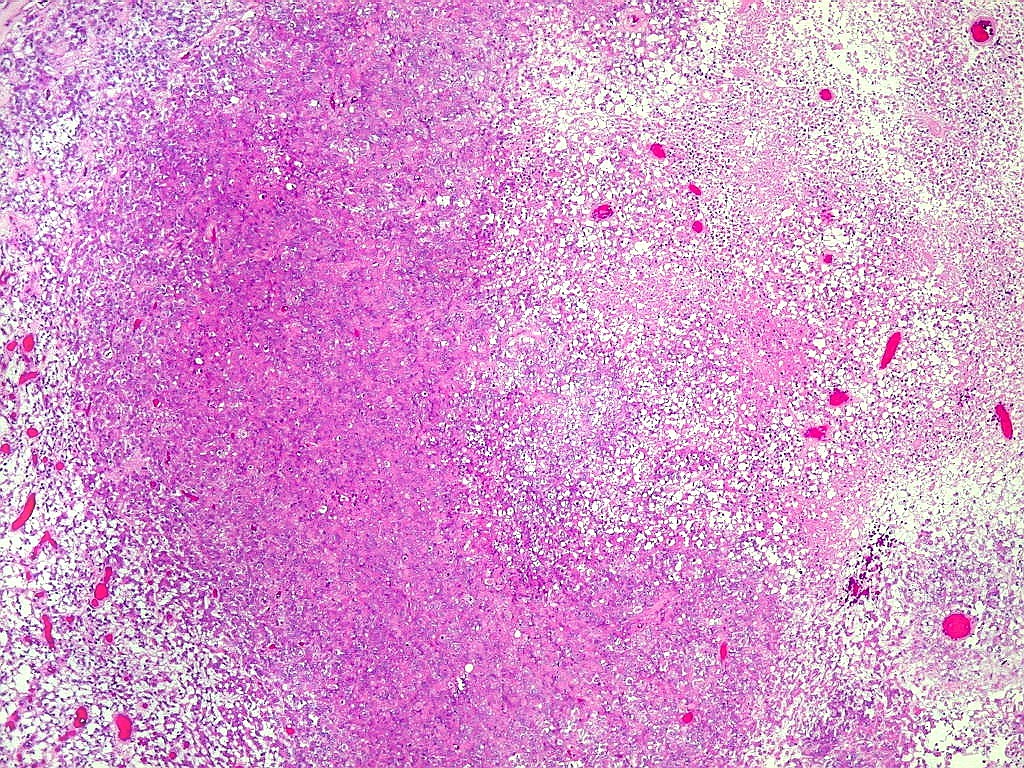
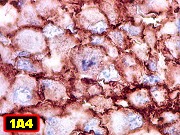
Microscopia.
Há amplo espectro de aparências histológicas, desde
tumores exclusivamente rabdóides a outros predominantemente de células
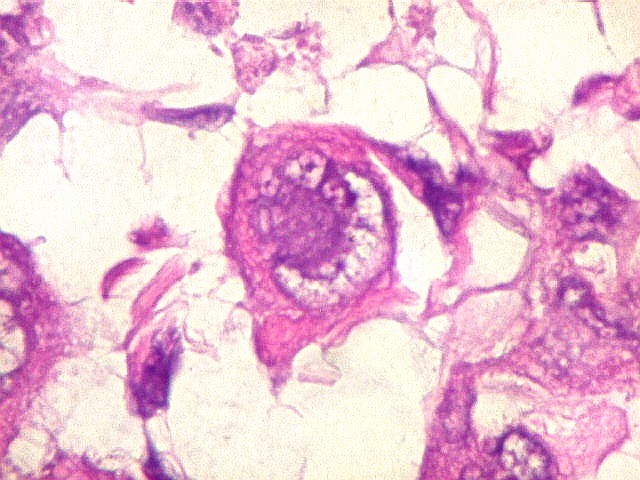
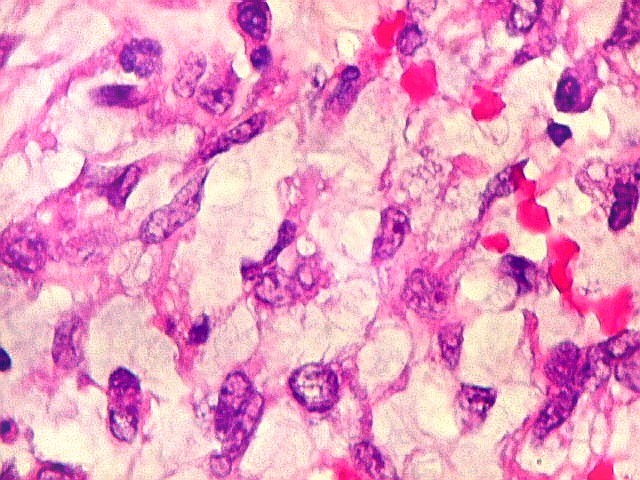
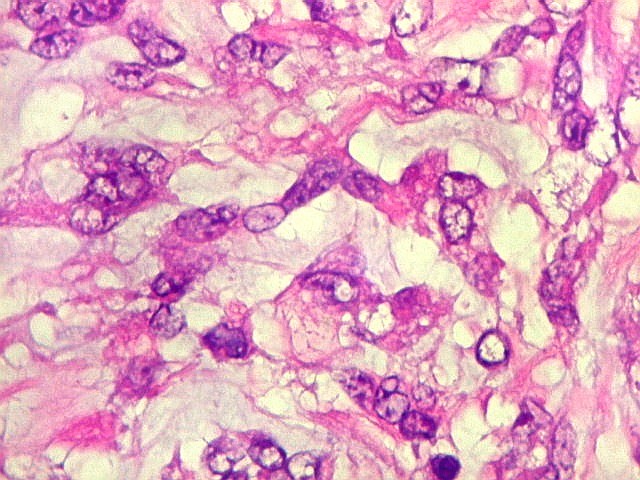
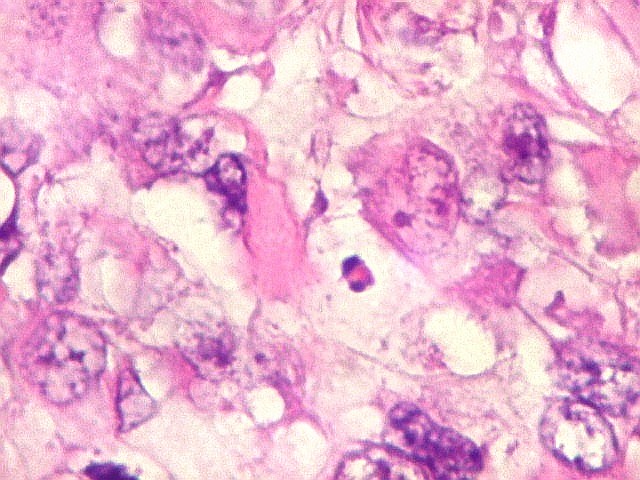
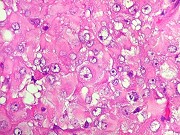
pequenas ou com componente de células claras. As células
rabdóides são geralmente grandes, com núcleo excêntrico
deslocado lateralmente por um enovelado de filamentos intermediários
preenchendo parte do citoplasma. O limite celular é bem definido.
O núcleo é grande, com aspecto vesiculoso e nucléolo
proeminente. As células grandes não são necessariamente
rabdóides. Possuem núcleos reniformes e amplo citoplasma
róseo, freqüentemente com vacuolização artefatual.
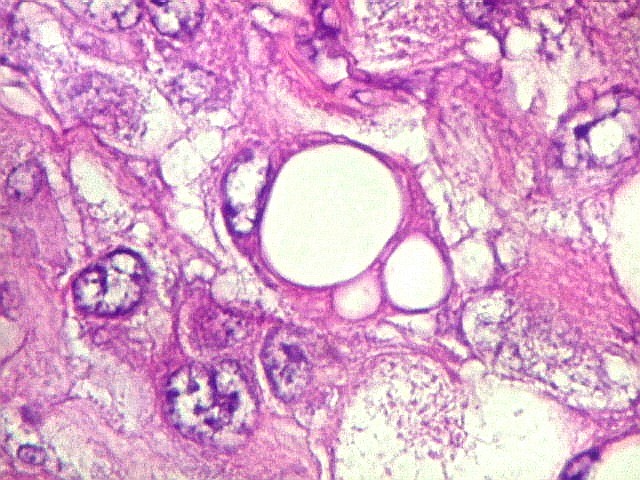
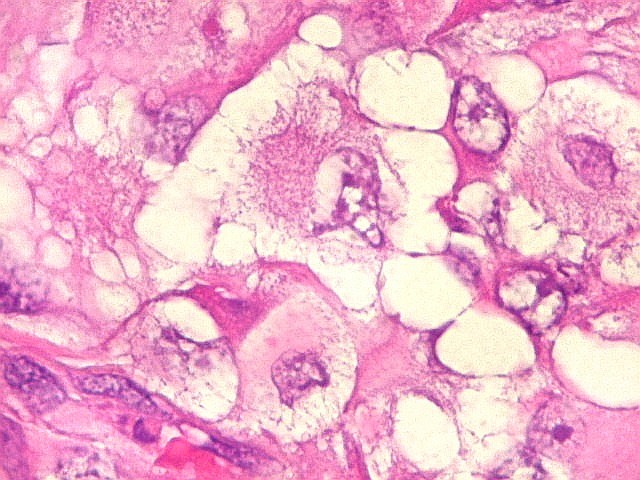
Em outras áreas pode haver células fusiformes em arranjo
fascicular. Quando há predomínio das células pequenas
a semelhança com meduloblastoma pode ser notável. Mais
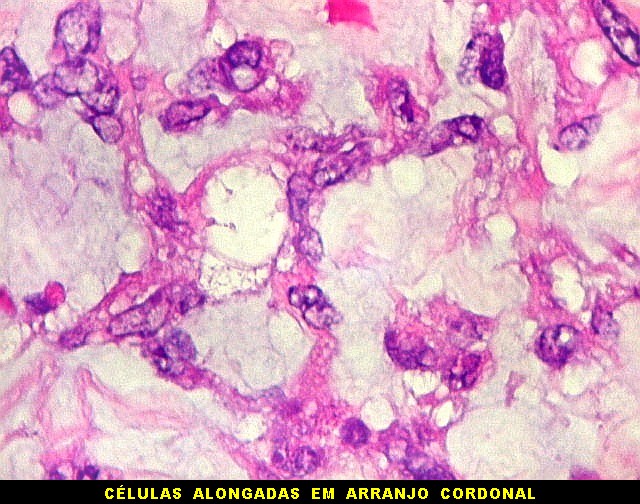
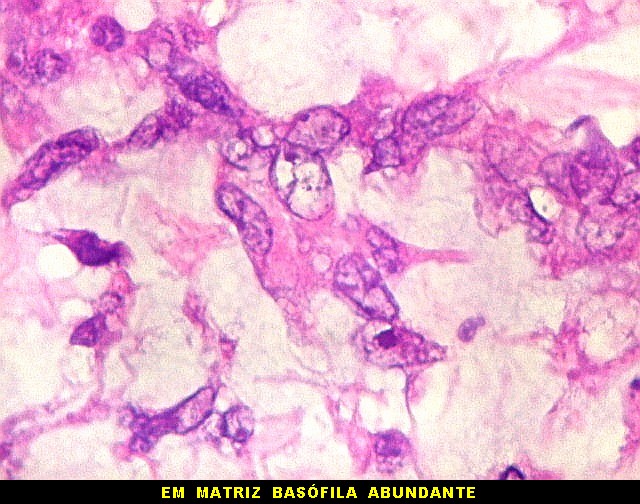
raramente, as células tomam arranjo cordonal em meio a matriz mucóide
basofílica, lembrando o padrão trabecular dos tumores rabdóides
renais. Em outros padrões mais raros, pode haver rosetas de
Flexner-Wintersteiner ou diferenciação epitelial na forma
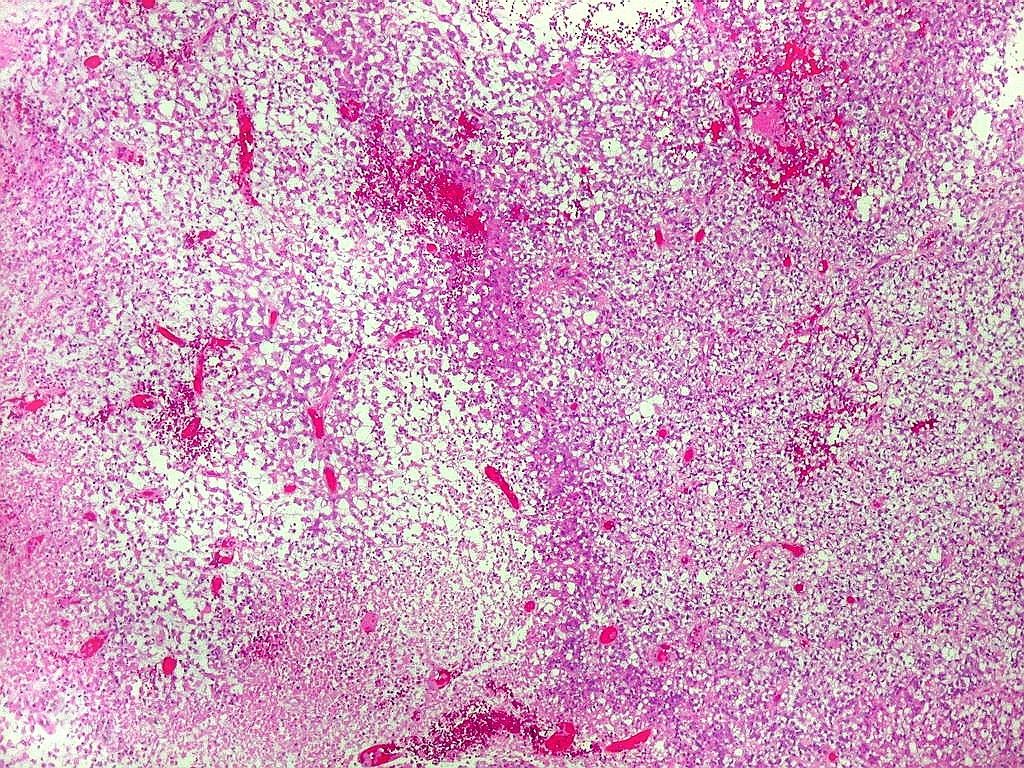
de glândulas. Necrose, freqüentemente com calcificação
distrófica, é comum. O aspecto heterogêneo
ajuda na diferenciação com os meduloblastomas, que são
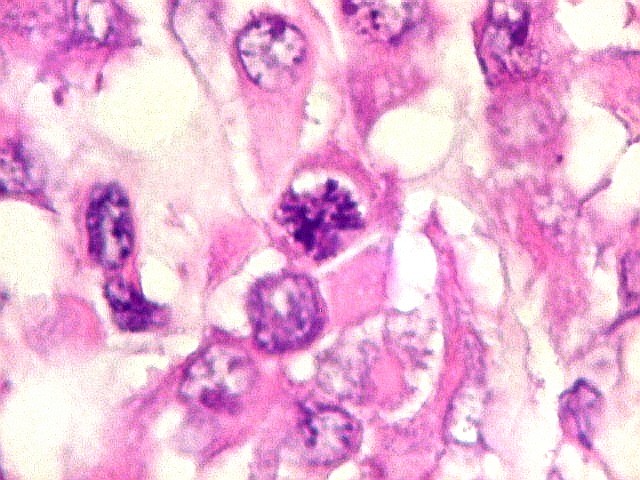
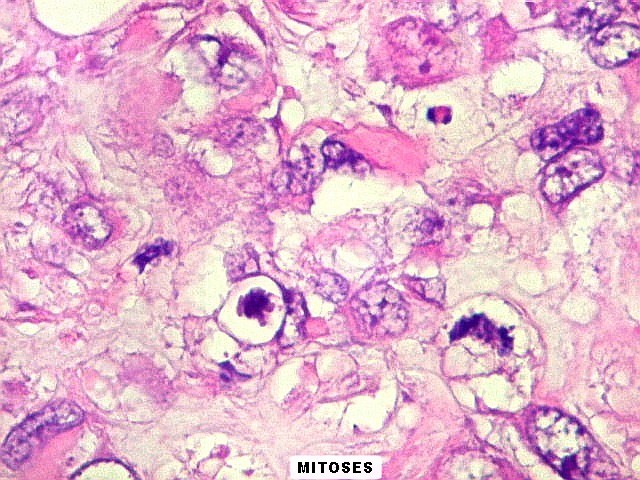
mais homogêneos, compactos e densamente celulares. Figuras
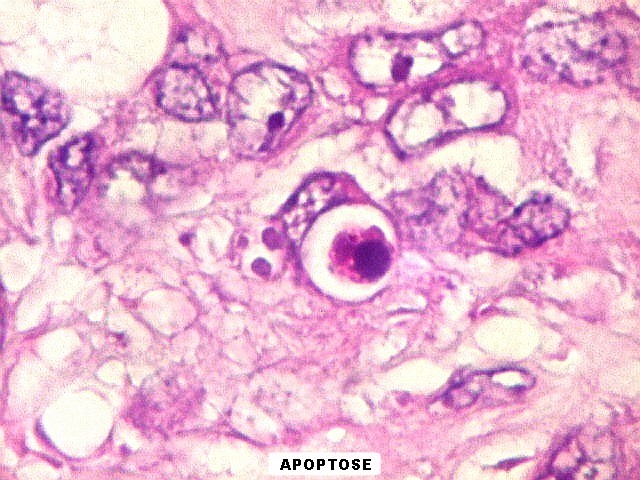
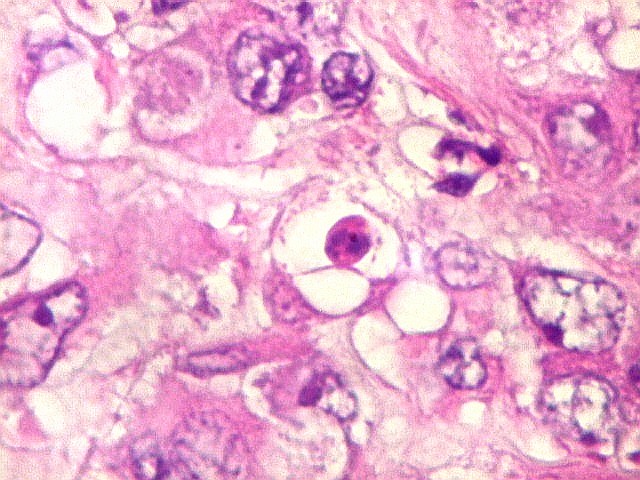
de mitose e apoptose são abundantes.
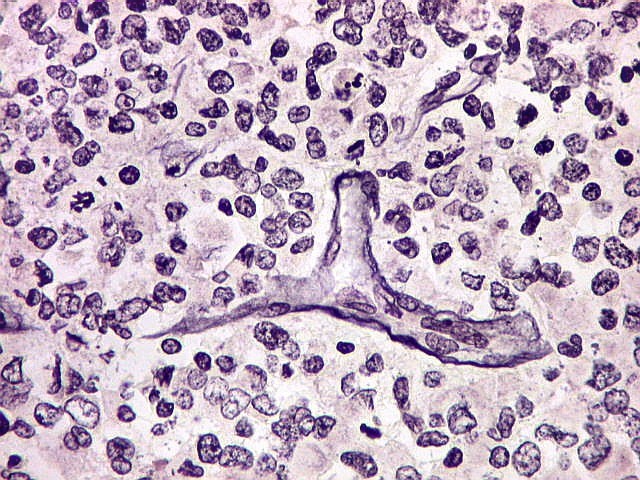
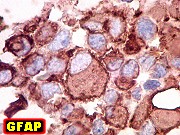
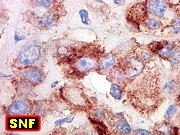
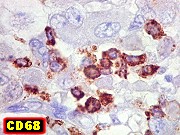
Imunohistoquímica.
O imunofenótipo é amplo, com as células grandes e
pálidas mostrando notável espectro de reatividade para diversos
antígenos. Positividade para vimentina é universal. Células
imunorreativas freqüentemente estão lado a lado a negativas.
Há positividade superficial nas células para EMA, o que é
de particular valia no diagnóstico diferencial com meduloblastomas.
Há também freqüente positividade para GFAP, citoqueratinas,
e menos comumente para actina de músculo liso, neurofilamento, sinaptofisina
e cromogranina. Células em fascículos são mais freqüentemente
positivas para actina. Marcadores de linhagem germinativa e músculo
esquelético são geralmente ausentes. Índice Ki67 é
acima de 50%.
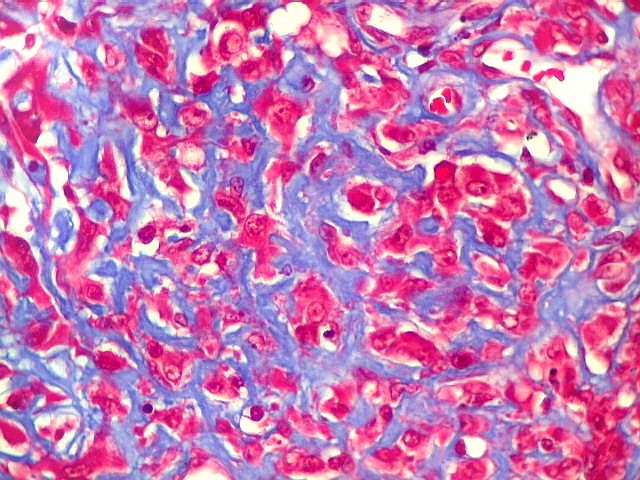
Microscopia eletrônica.
Células rabdóides mostram no citoplasma enovelados densos
de filamentos intermediários.
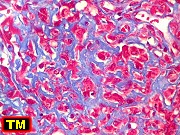
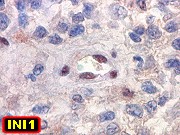
Genética molecular.
Na grande maioria dos ATRTs há perda de todo ou parte do cromossomo
22. Deleções e mutações no gene INI1
em 22q.11.2 parecem ser centrais no desenvolvimento de tumores rabdóides
renais e extrarenais. Esta pode ser detectada por FISH ou imunohistoquímica
para proteína INI1, que será negativa (ausente) nos núcleos
das células neoplásicas. A demonstração
da inativação de INI1 é condição sine
qua non para o diagnóstico de ATRT. Casos com esta morfologia,
mas sem confirmação desta alteração, devem
ser classificados como tumor embrionário do sistema nervoso central
com feições rabdóides. INI1 (ou SMARCB1)
é uma proteína nuclear expressa constitucionalmente em células
normais e na maioria dos tumores. Funciona em dependência de ATP
na modelação da estrutura da cromatina. Há casos familiais,
nas chamadas síndromes de predisposição a tumores
rabdóides.
Diagnóstico diferencial.
O principal é o meduloblastoma, comentado acima. Também
astrocitomas gemistocíticos podem causar confusão, pela semelhança
destes astrócitos com as células rabdóides. A IH positiva
para EMA ajuda. Casos com aspecto epitelióide podem lembrar carcinomas
do plexo coróide, que são na maioria supratentoriais. Carcinomas
do plexo são EMA negativos e citoqueratina positivos.
Célula de origem
é desconhecida, mas devido à diferenciação
divergente e ocorrência em crianças pequenas, propõe-se
derivação de células fetais pluripotentes.
Tratamento e prognóstico.
Sendo volumosos e profundos, é difícil conseguir-se ressecção
completa. Radioterapia não é aconselhada devido à
tenra idade dos pacientes. Opta-se por quimioterapia agressiva, mas a sobrevida
é geralmente da ordem de 6 meses a 1 ano. Contudo, estudos
multiinstitucionais recentes demonstraram sobrevida mais animadora.
Fontes.
Burger,
PC, Scheithauer BW, Vogel FS. Surgical Pathology of the Nervous System
and its Coverings. 4th Ed. Churchill Livingstone, New York, 2002.
pp 336-9.
Judkins
AR et al. Atypical teratoid/rhabdoid tumour. In
WHO Classification of Tumours of the Central Nervous System. Revised
4th Ed. Louis DN et al. (eds). International Agency for Research
on Cancer, Lyon, 2016. pp 209-12. |