|
|
compatível com linfoma de zona marginal extranodal |
|
|
|
compatível com linfoma de zona marginal extranodal |
|
| Esta lesão inicialmente foi interpretada na HE como infiltrado inflamatório crônico inespecífico, compatível com pseudotumor inflamatório em vista dos estudos de imagem e da boa evolução com corticoterapia. Como a topografia e características de imagem fossem fortemente sugestivas de meningioma, levantou-se também a hipótese de um meningioma rico em linfócitos e plasmócitos. A imunohistoquímica foi pedida para investigar esta possibilidade, mas EMA e AE1AE3 foram negativos. Por outro lado, os marcadores para células hematológicas definiram o caso como um raro linfoma não Hodgkin da base do crânio, de imunofenótipo B e baixo grau histológico, melhor interpretado como linfoma de zona marginal extranodal, colocando-se diagnóstico diferencial com linfoma linfoplasmocítico. Como este diagnóstico só foi firmado num reestudo do caso, dois anos após a biópsia e depois do óbito da paciente por outras causas, não dispomos de dados clínicos ou laboratoriais que possibilitassem melhor caracterização do ponto de vista hematológico. |
| Destaques da microscopia. | ||
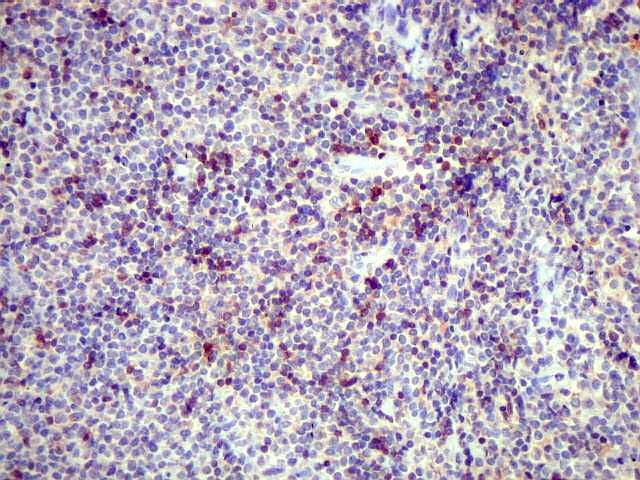
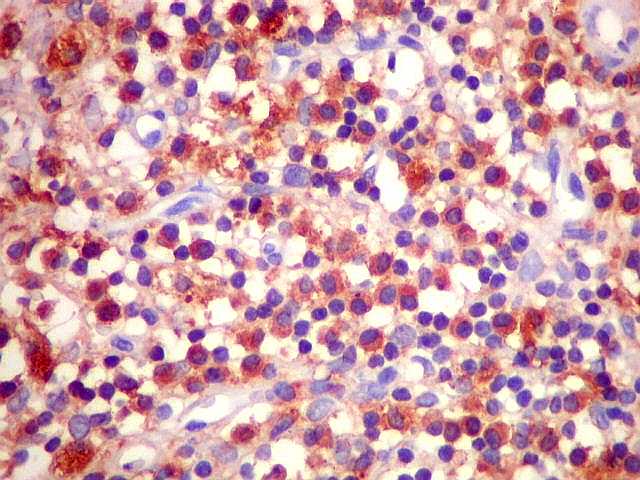
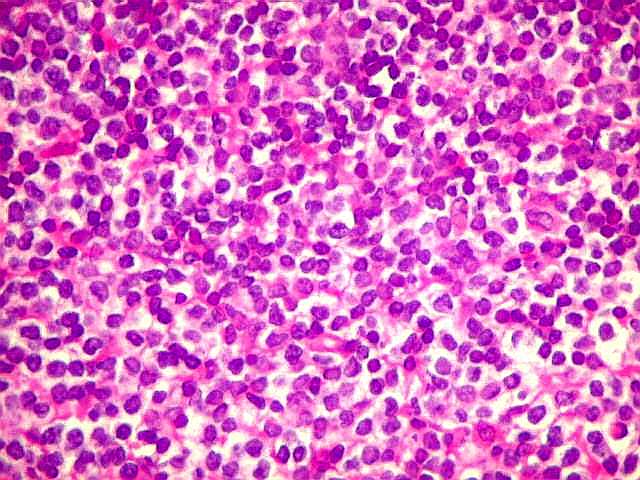
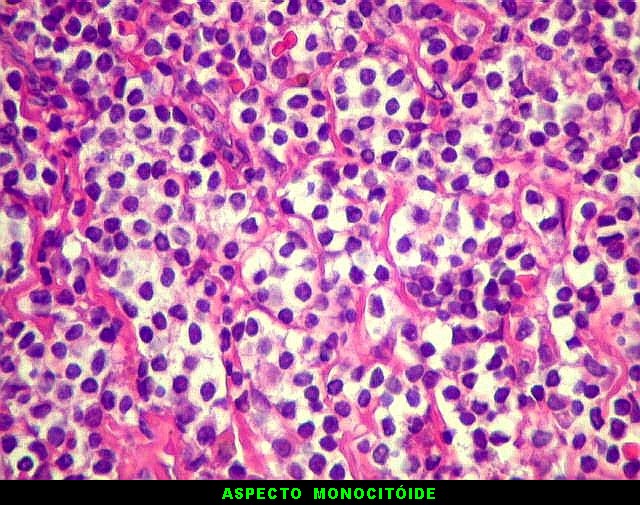
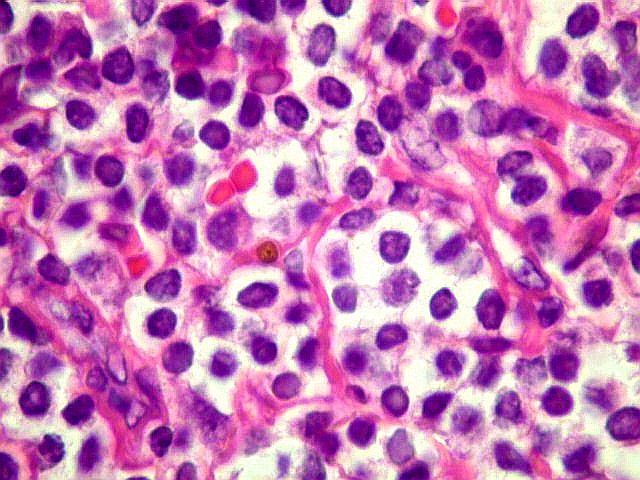
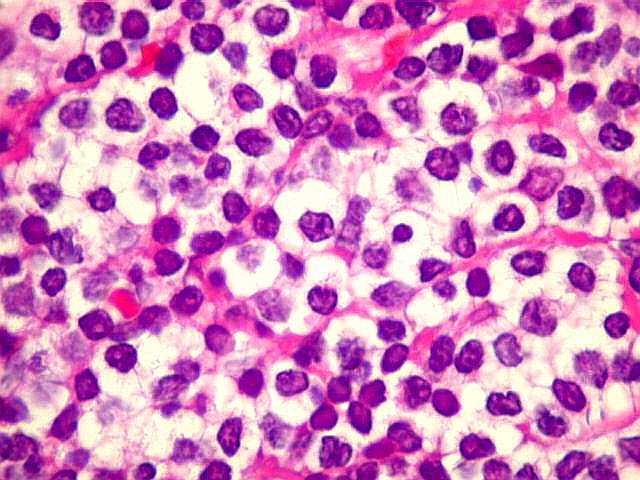
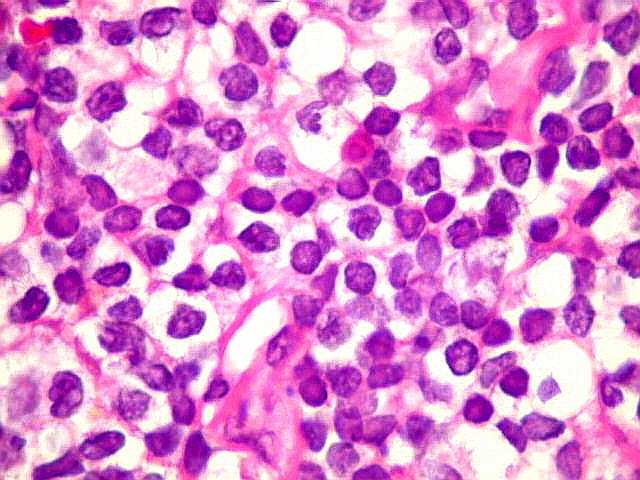
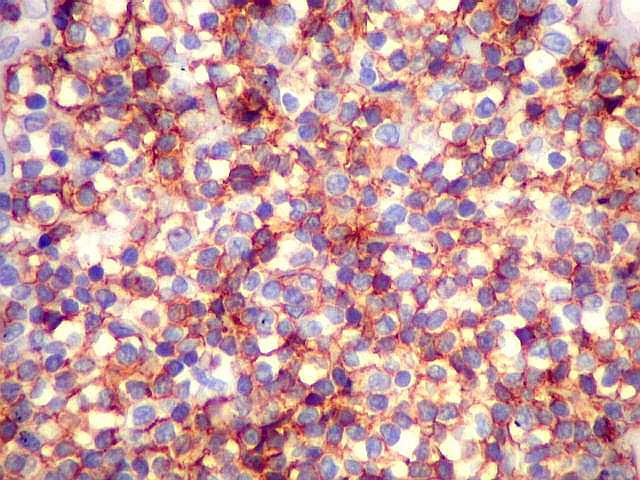
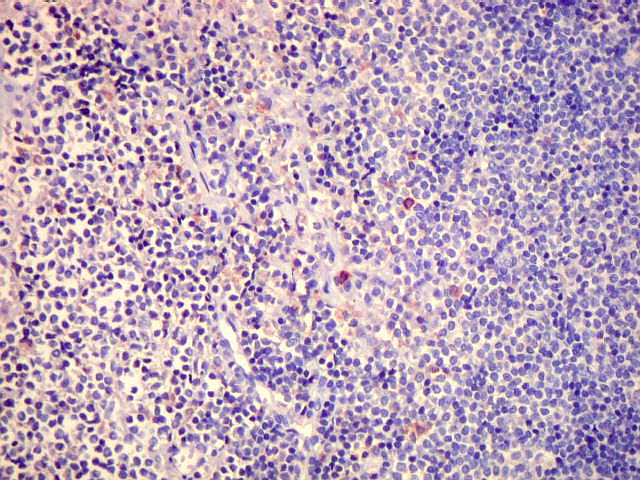
| HE. Caráter monótono, predomínio de pequenos linfócitos, com citoplasma amplo (aspecto monocitóide). | Plasmócitos, células plasmocitóides. | CD20. Positividade difusa nos linfócitos B tumorais. |
 |
 |
 |
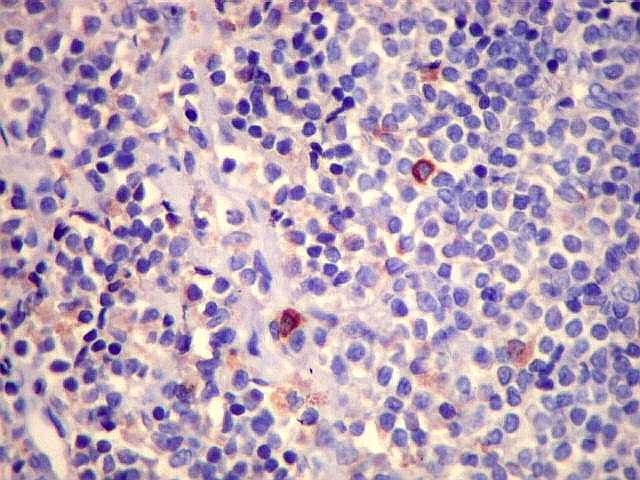
| CD3. Positividade em pequenos linfócitos T esparsos. | CD5. Negativo nas células tumorais (esperado); positivo em linfócitos T esparsos. | CD68. Positivo em macrófagos. |
 |
 |
 |
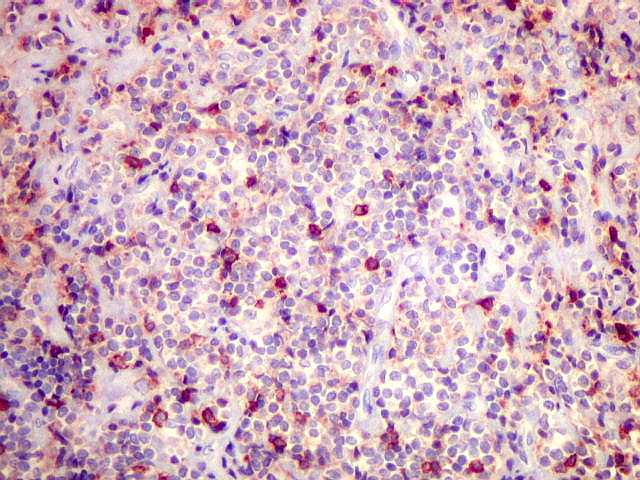
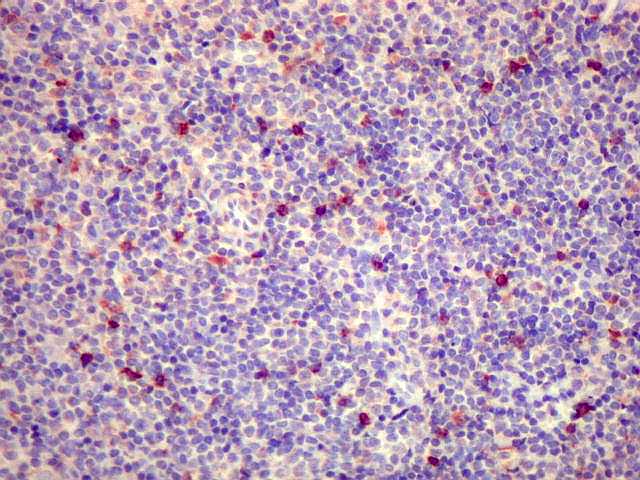
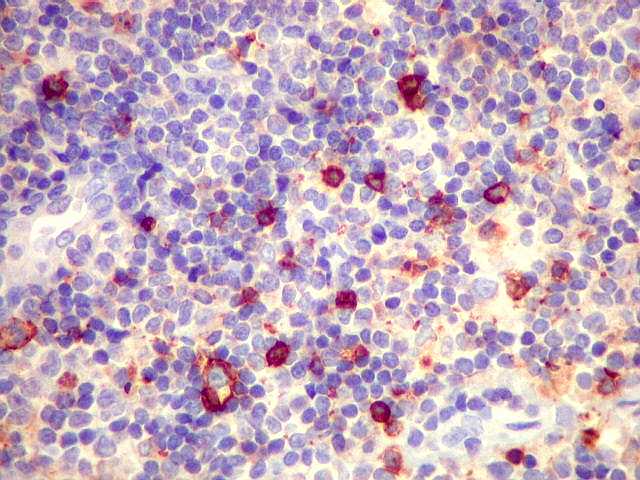
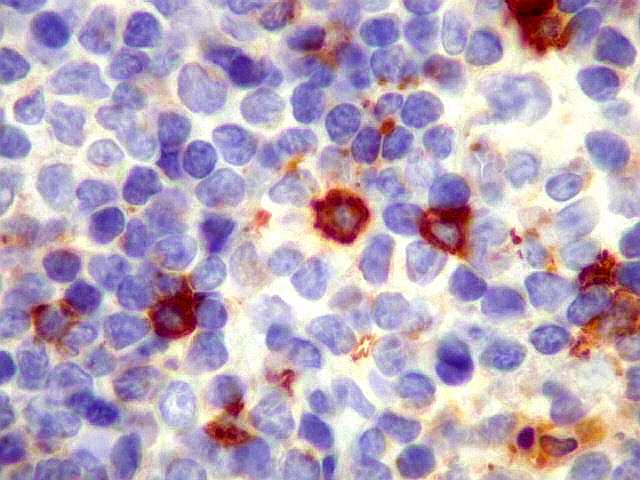
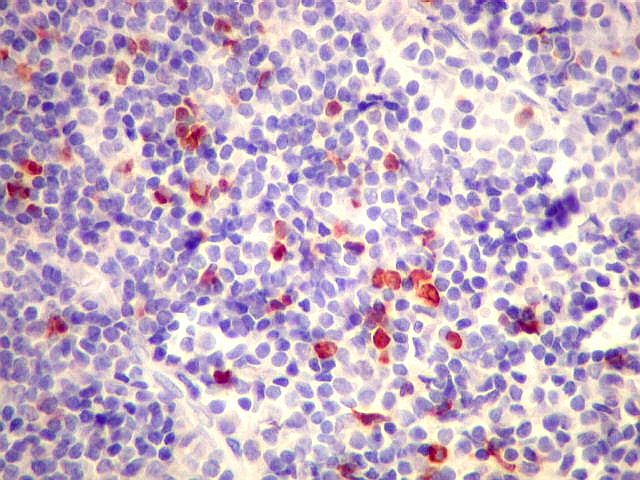
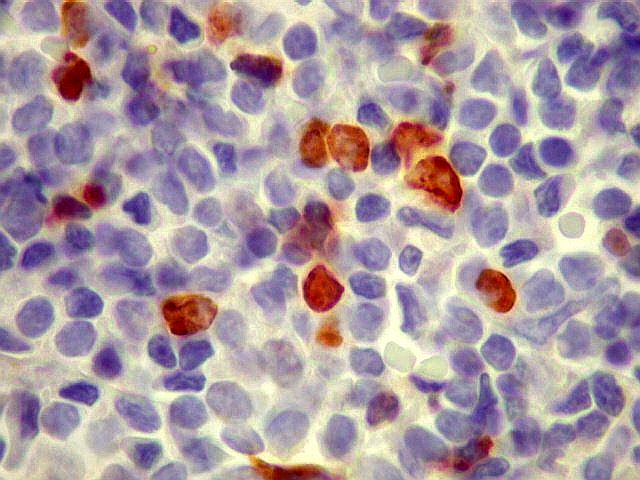
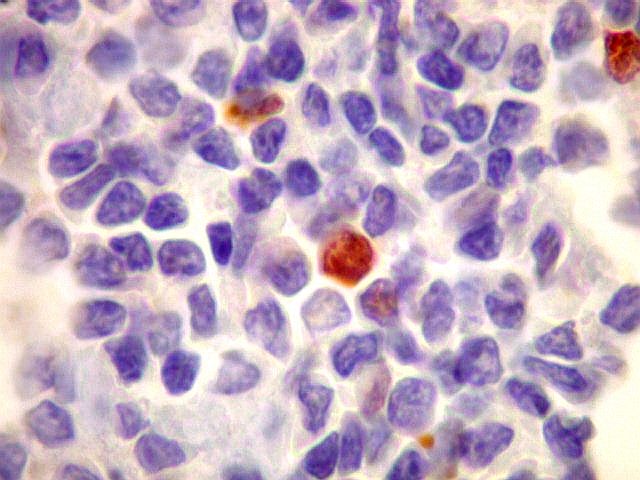
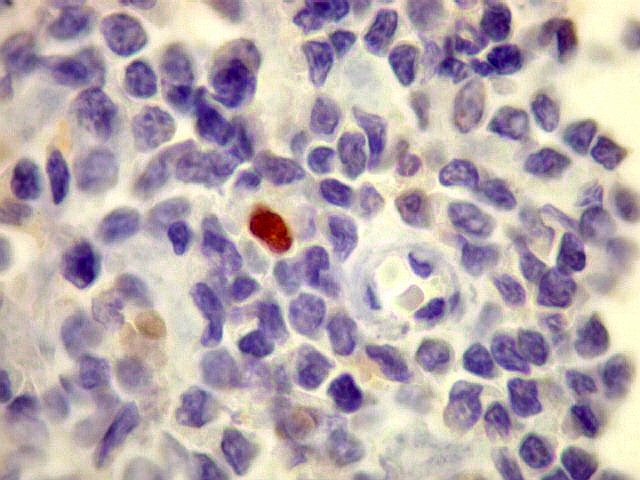
| kappa. Cadeia leve, positiva em plasmócitos no tumor. | lambda. Cadeia leve, negativa em plasmócitos no tumor (restrição de cadeia leve). Positiva em alguns plasmócitos residuais. | Ki-67. Positividade em cerca de 3% dos núcleos das células neoplásicas. |
 |
 |
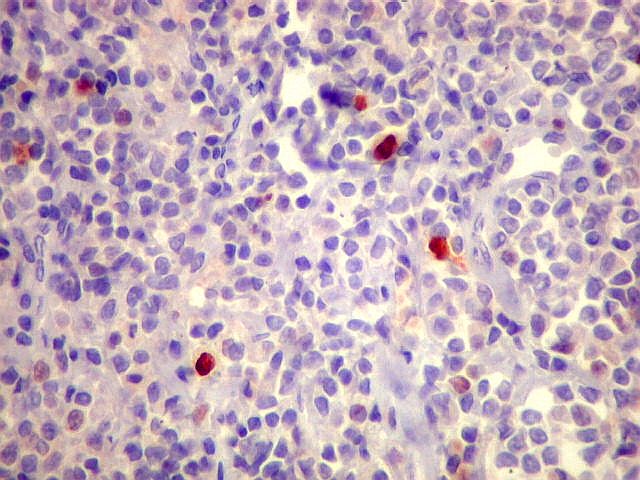 |
| Obs. Negativo também para CD56, CD57, BCL-1, AE1AE3, EMA, S-100, p53. | ||
|
|
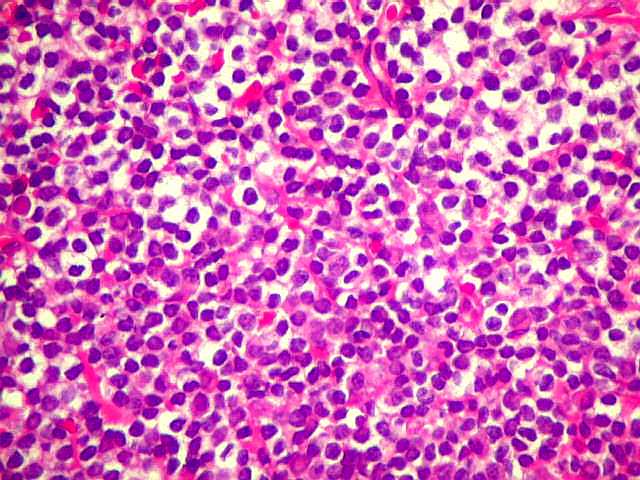
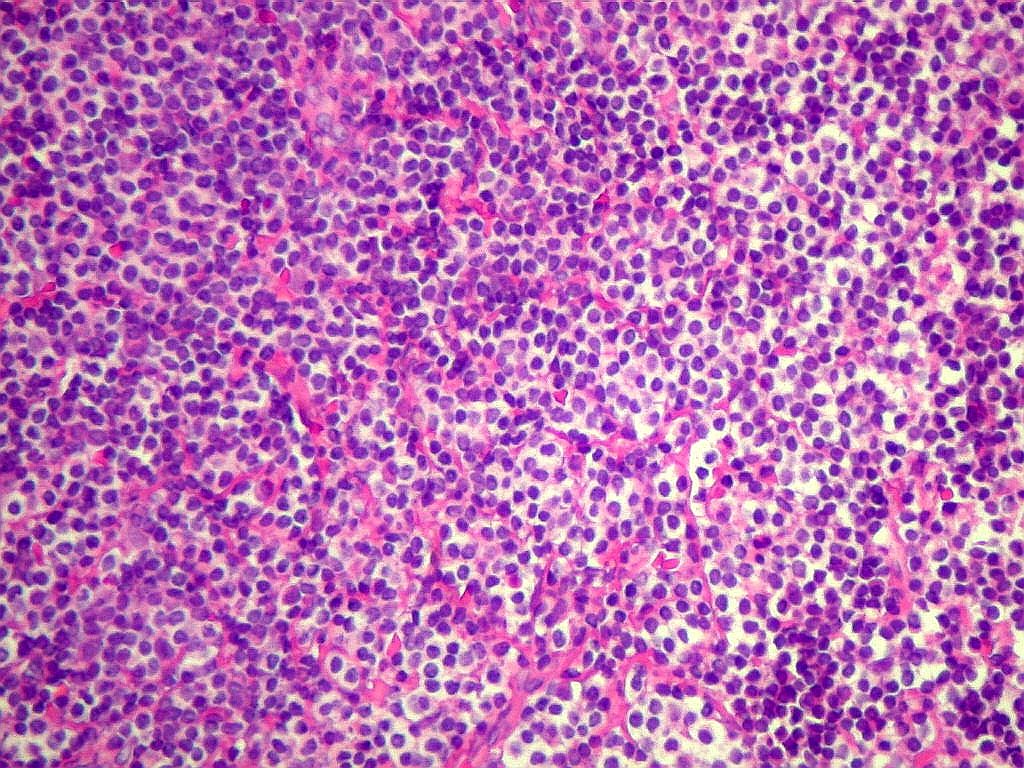
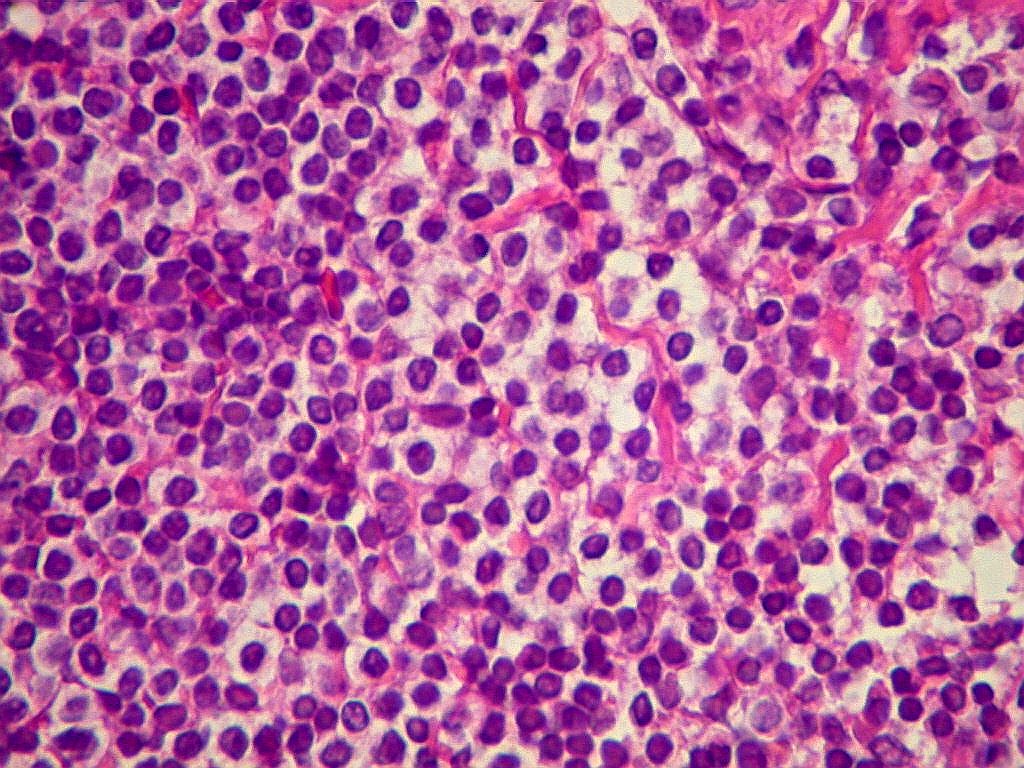
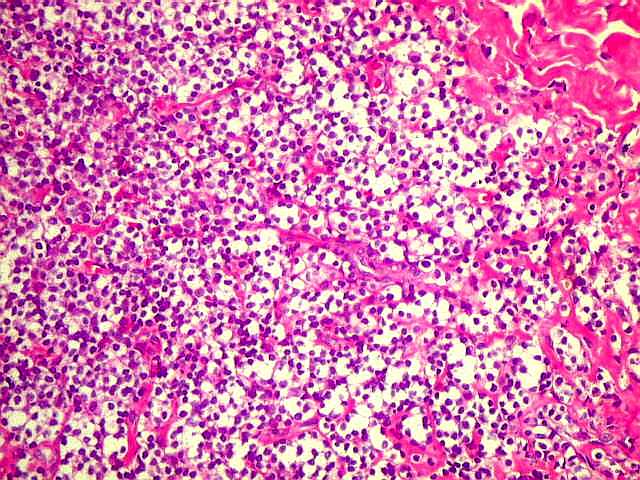
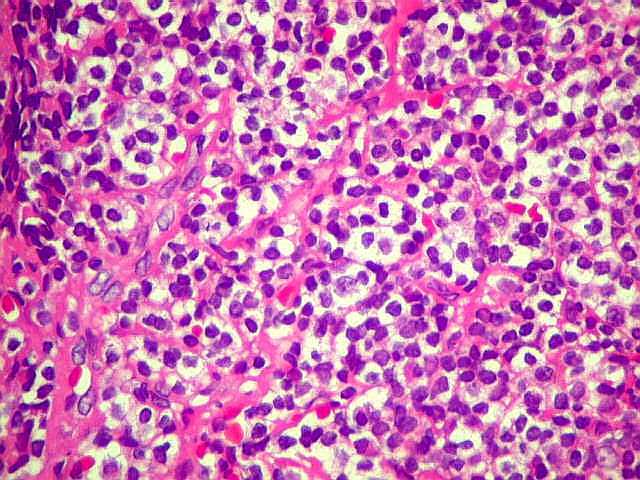
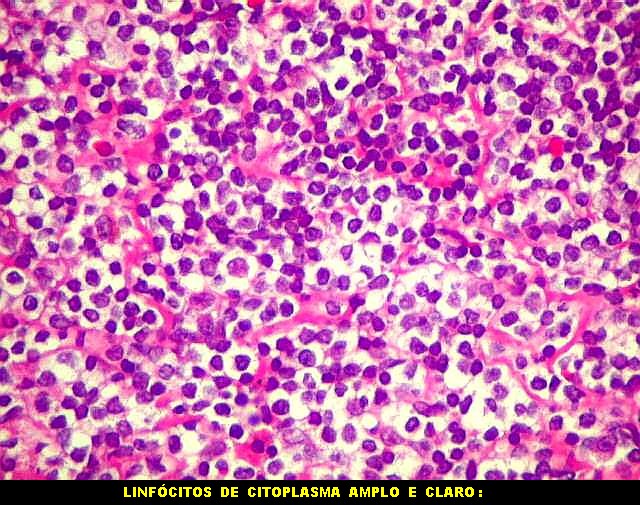
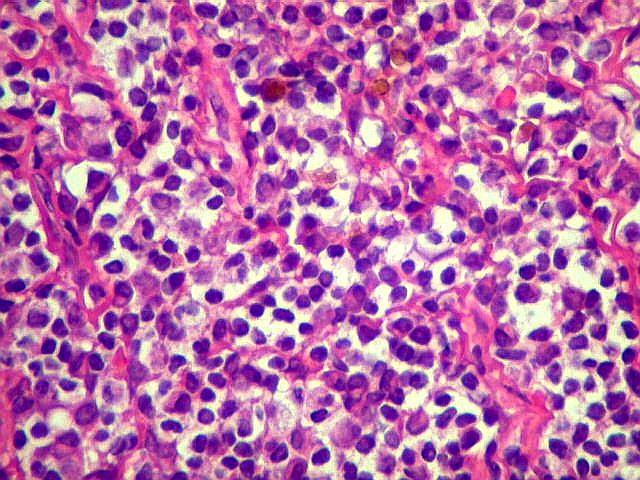
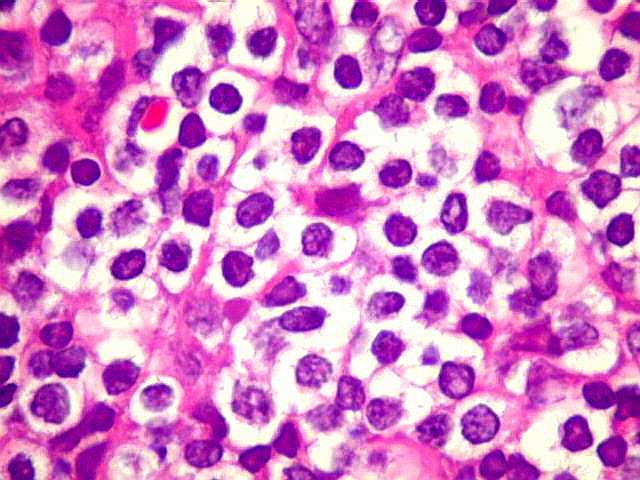
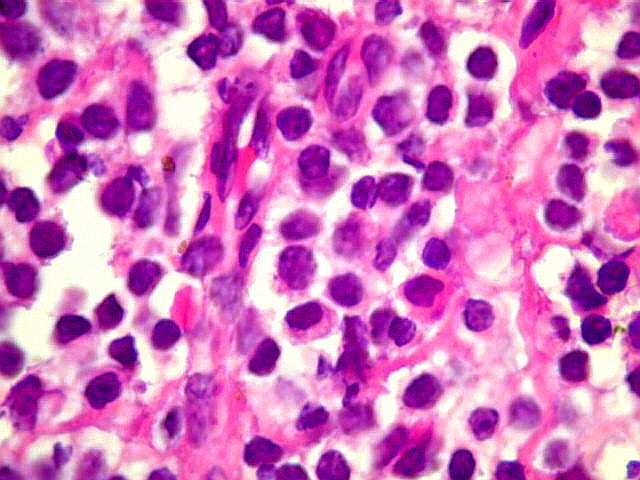
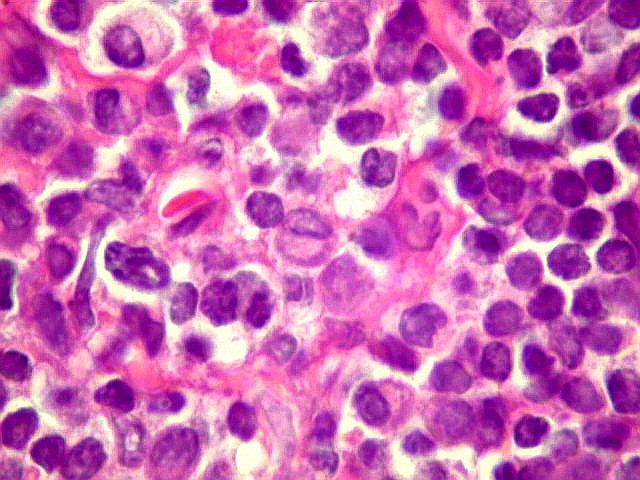
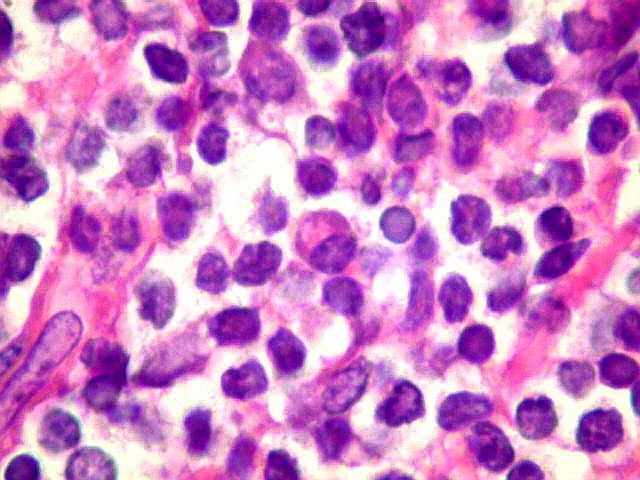
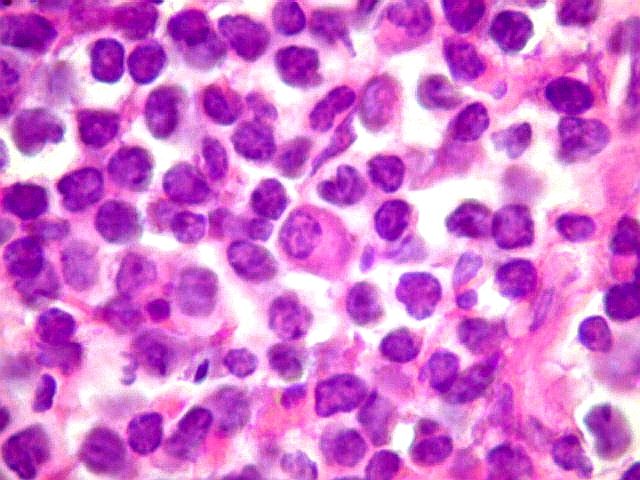
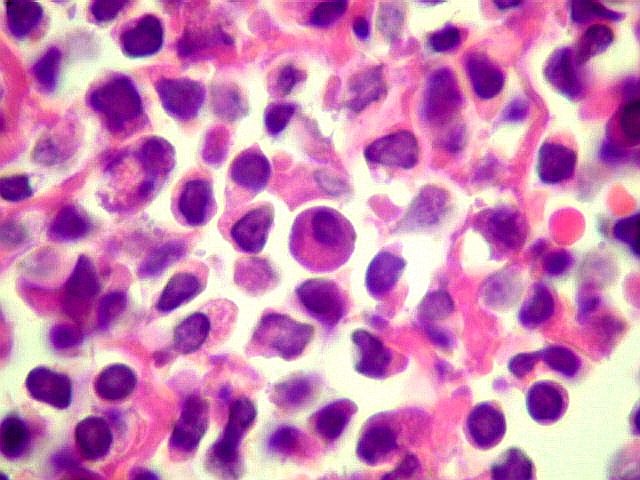
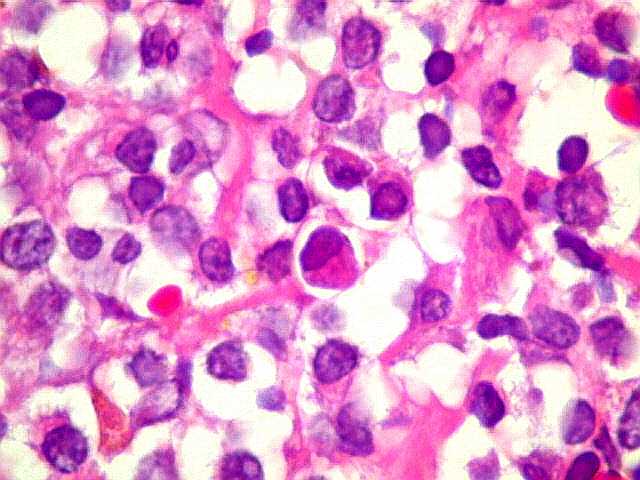
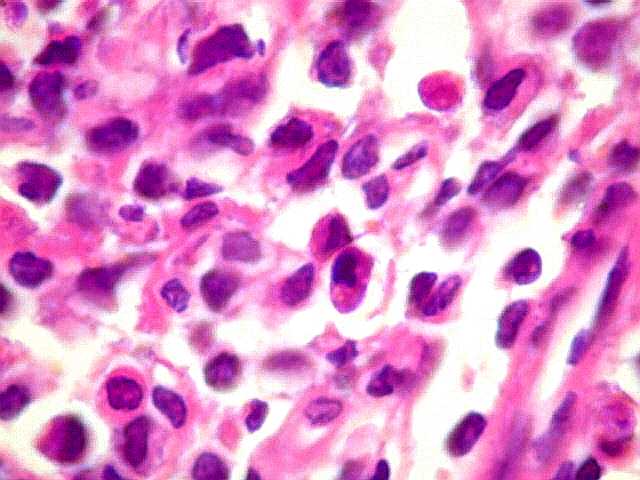
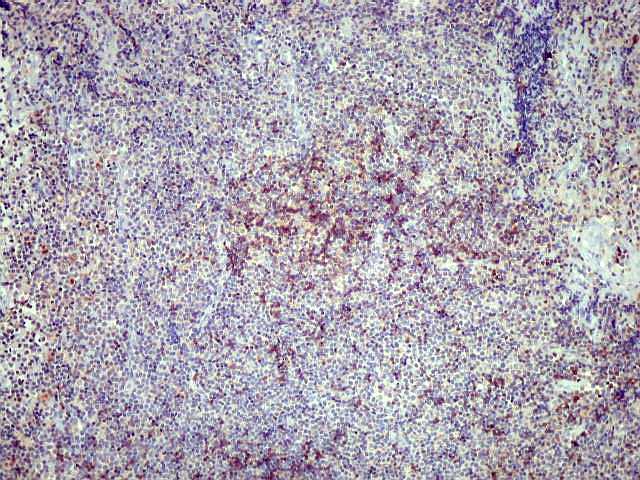
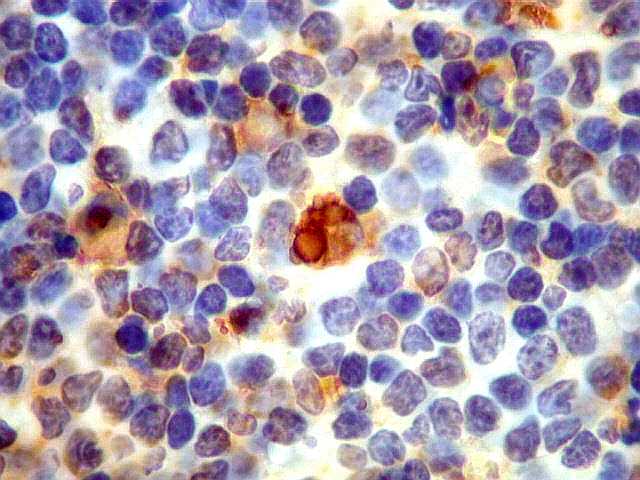
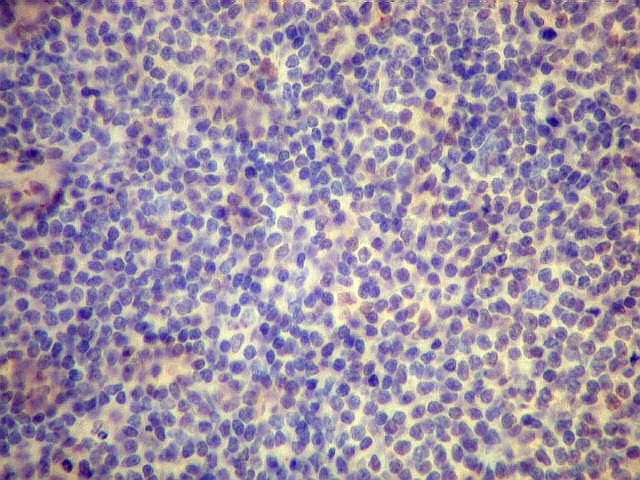
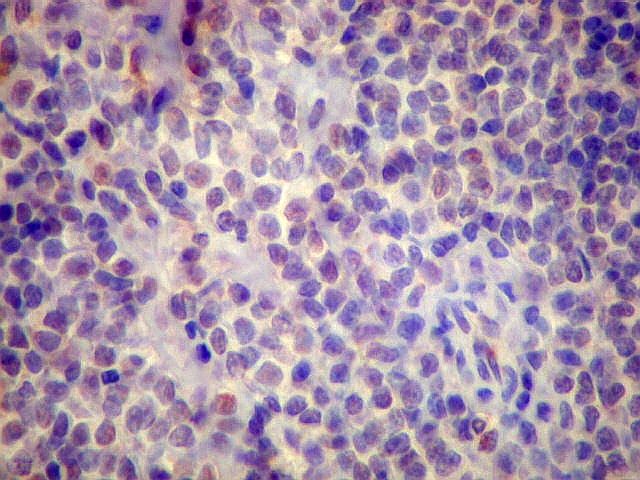
| Aspecto geral do tumor. A lesão é constituída basicamente por linfócitos densamente agrupados em arranjo sólido, com aspecto indolente, mostrando regularidade dos núcleos, que são na grande maioria pequenos, com cromatina densa. Em áreas, as células apresentam citoplasma amplo e claro, o que lhes confere um aspecto chamado de monocitóide. Há também plasmócitos e células assemelhadas (linfócitos plasmocitóides) esparsos, que induziram ao diagnóstico de pseudotumor inflamatório na primeira abordagem do caso, quando não foi realizada imunohistoquímica. Não há estruturas que lembrem folículos linfóides ou centros germinativos. Não foram observadas figuras de mitose. |
 |
 |
 |
 |
|
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
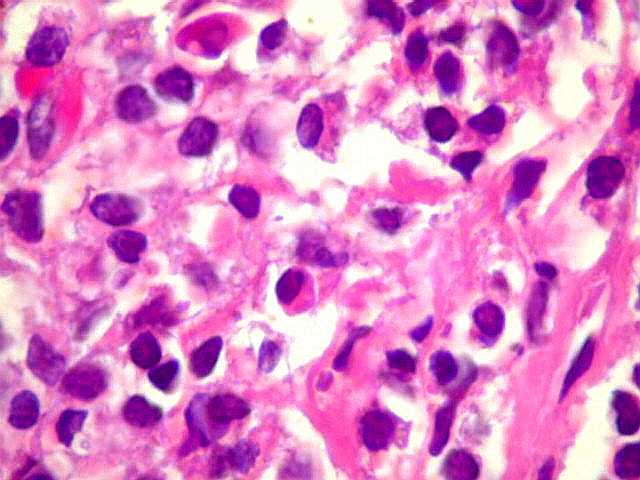 |
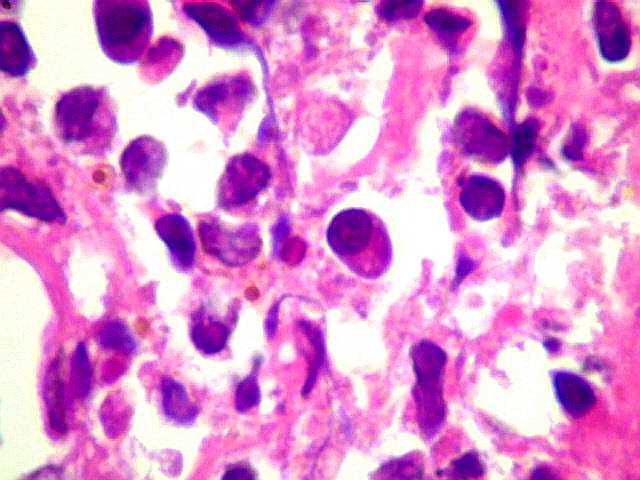
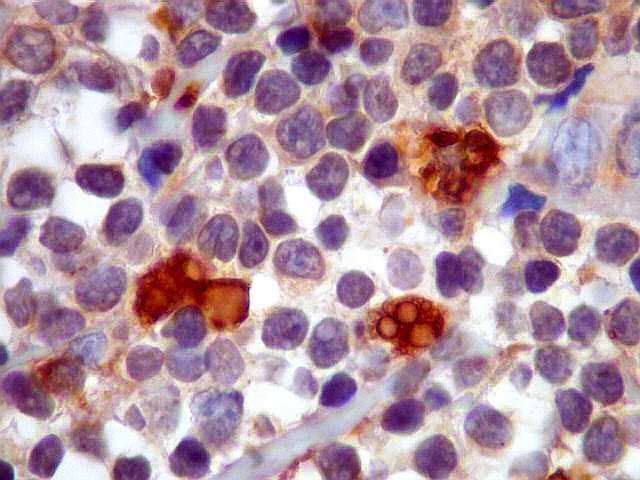
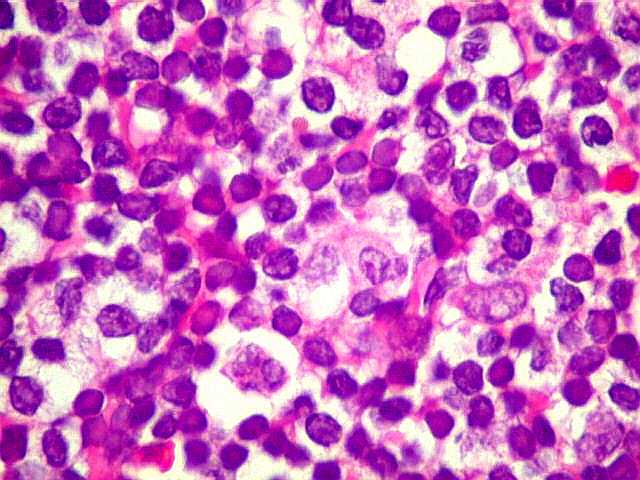
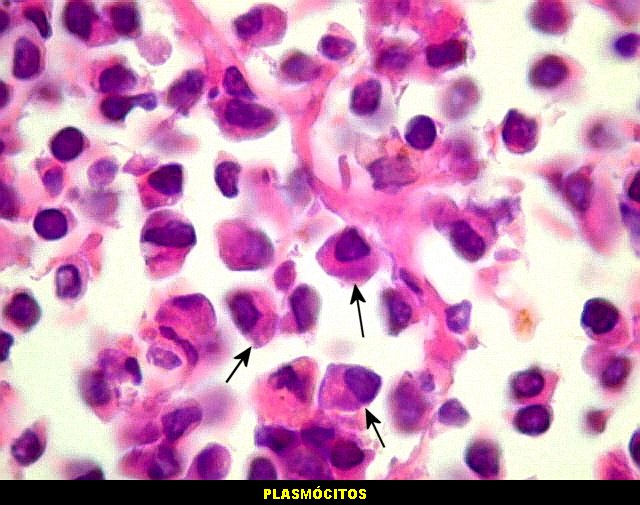
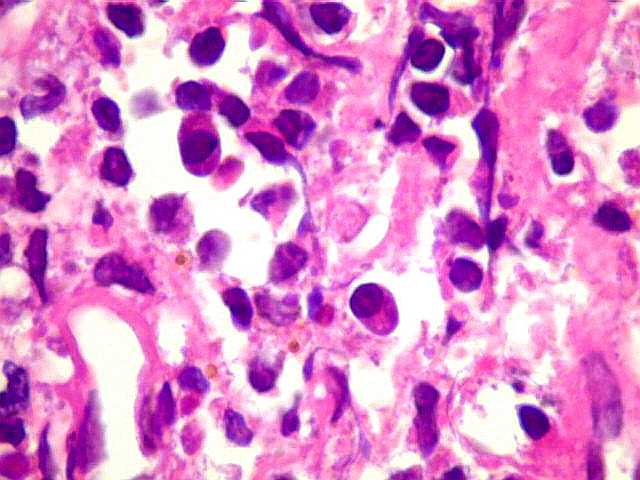
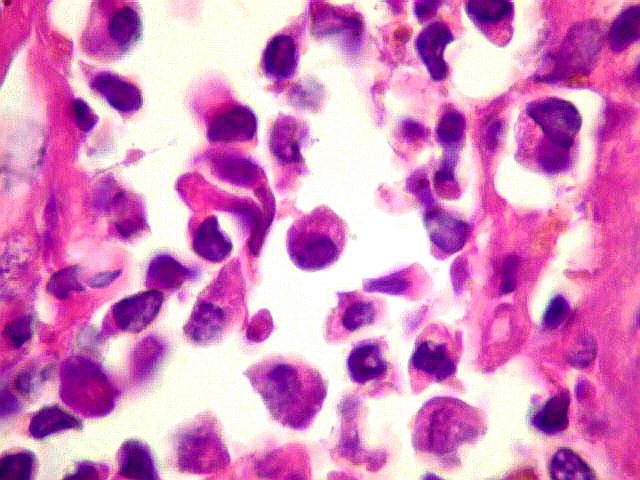
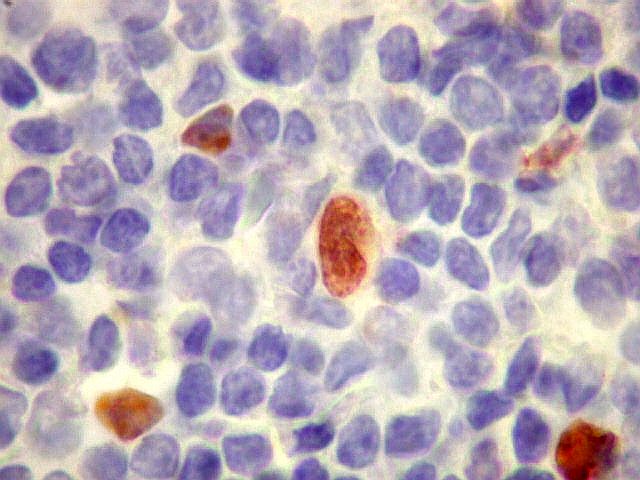
| Plasmócitos e células plasmocitóides. Em meio ao mar de linfócitos monótonos, eram observadas várias células com morfologia de plasmócitos bem diferenciados, com núcleo excêntrico e citoplasma de basofilia variável. | |
 |
 |
 |
 |
 |
 |
|
 |
 |
 |
|
|
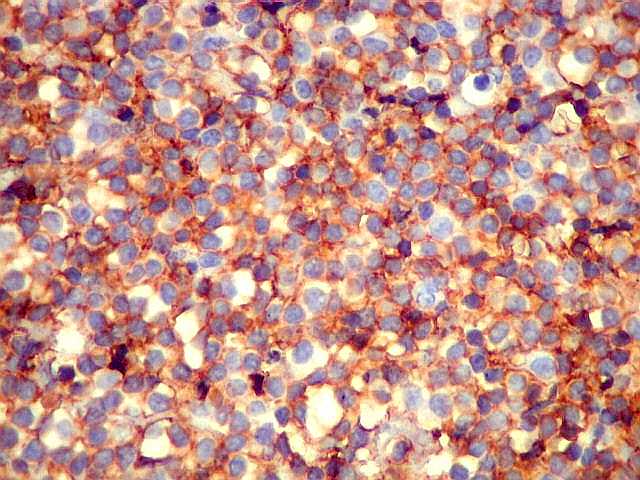
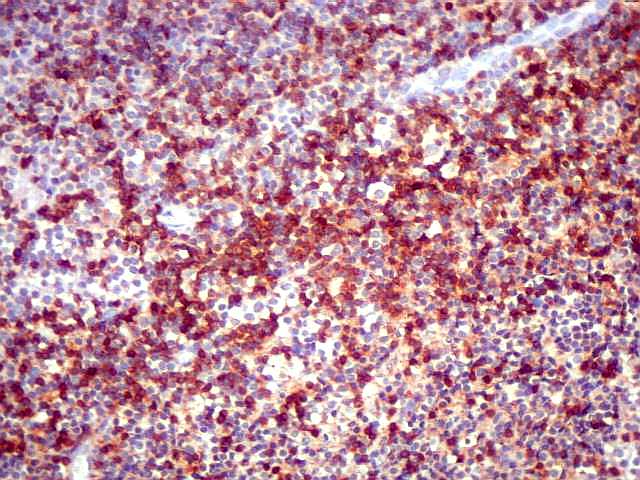
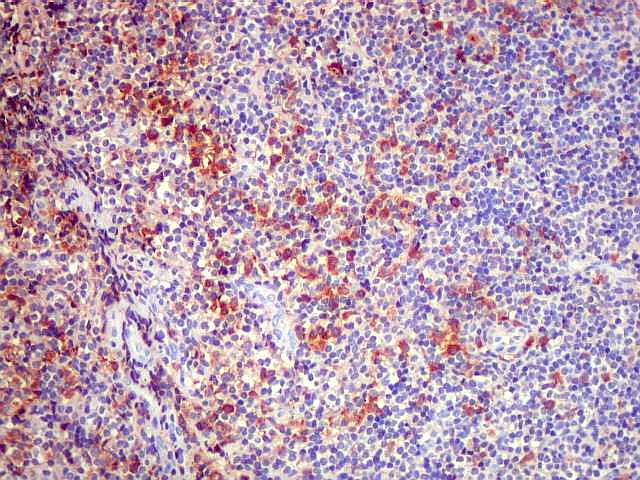
| CD20. Os linfócitos são em sua quase totalidade positivos para CD20, um marcador de linfócitos B. | |
 |
 |
|
 |
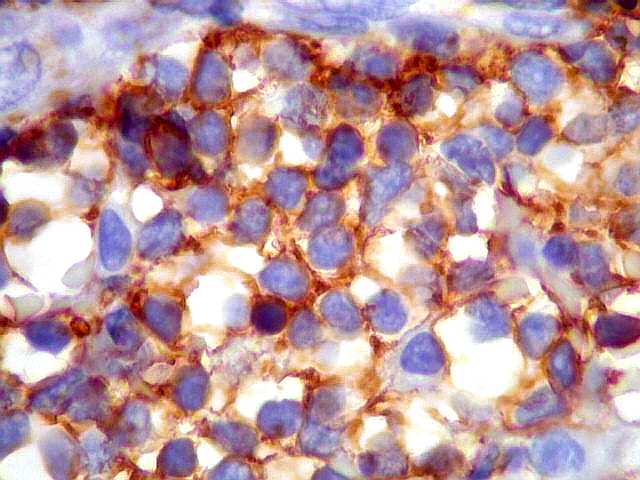 |
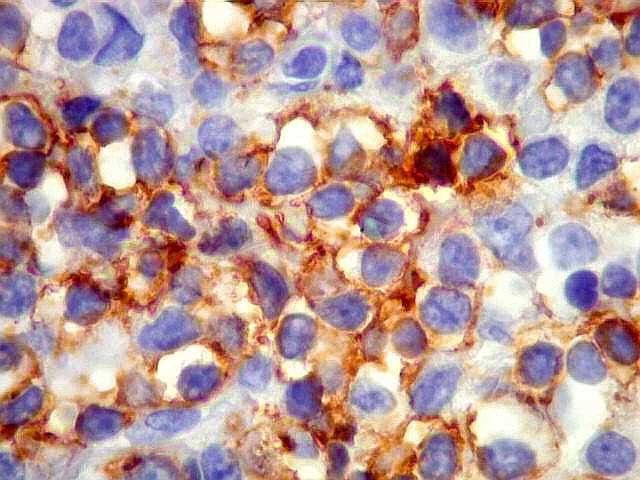 |
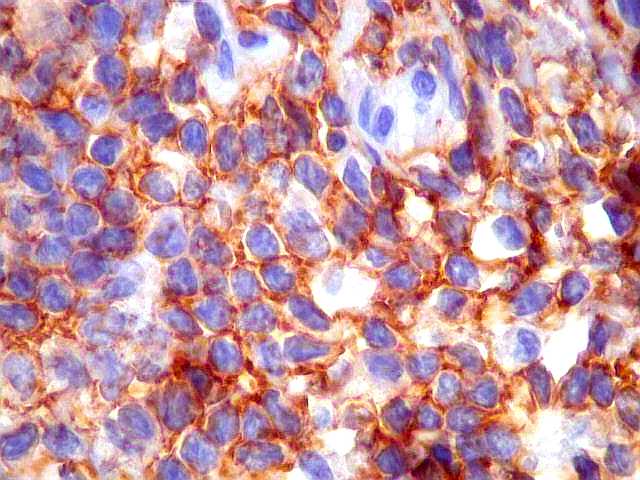 |
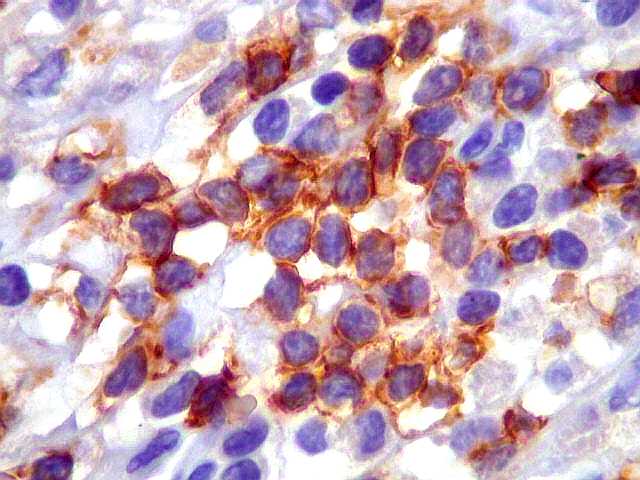 |
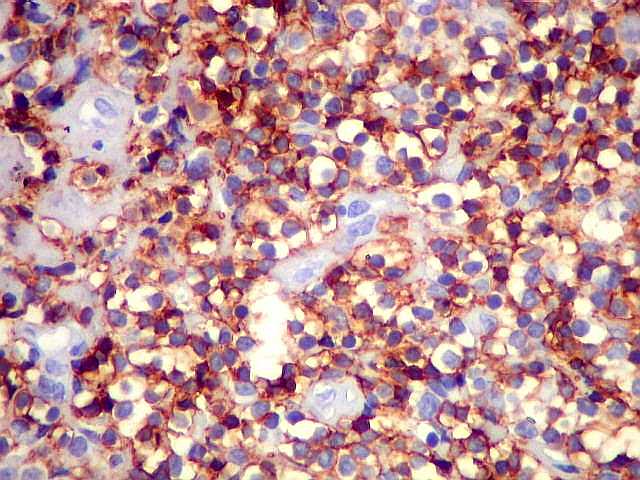
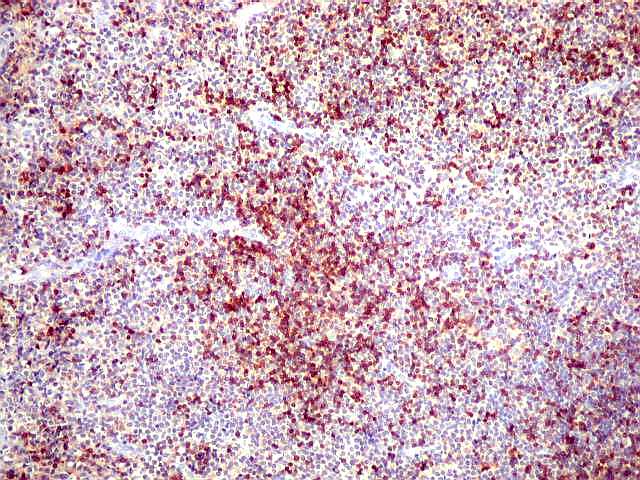
| CD3. Linfócitos T eram encontrados entre os muito mais numerosos linfócitos B, e são considerados reacionais ou préexistentes. Seus núcleos são menores que os dos linfócitos B neoplásicos. | |
 |
 |
 |
 |
 |
|
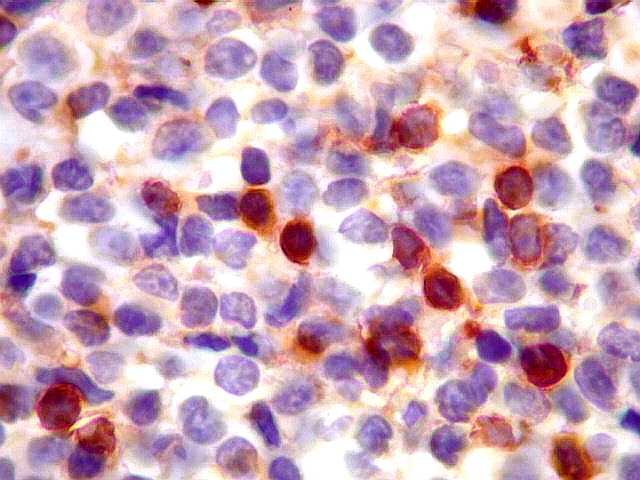 |
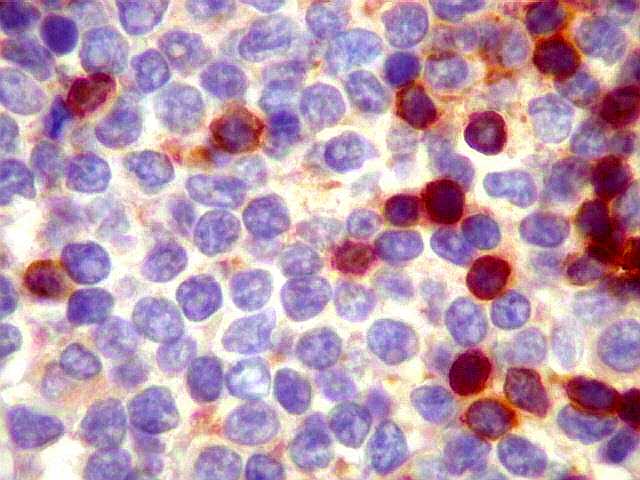 |
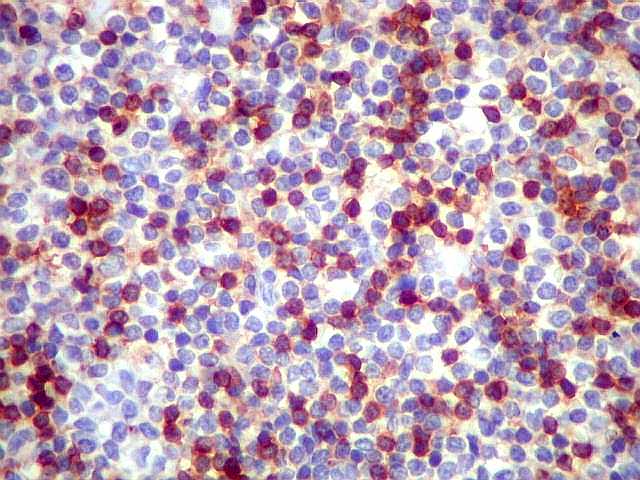
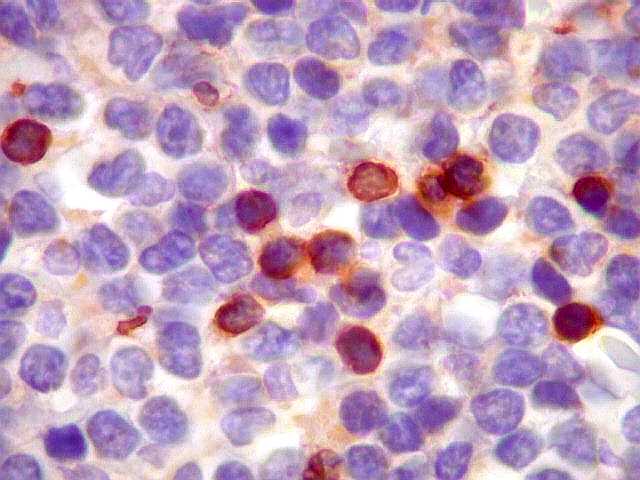
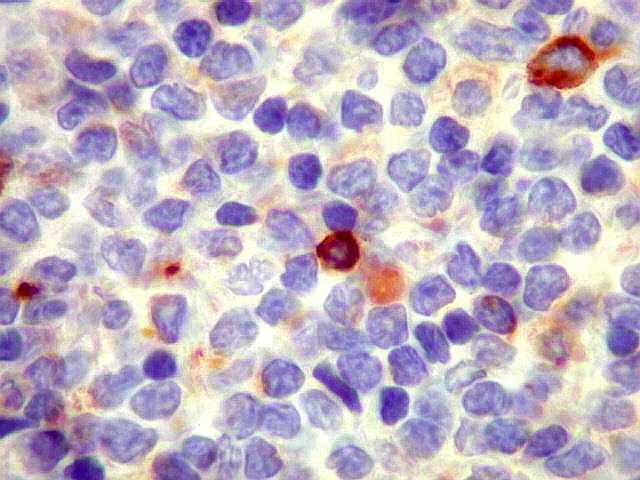
| CD5. Este marcador T é habitualmente negativo em linfomas de zona marginal e linfoplasmocíticos. As células marcadas são linfócitos T préexistentes. CD5 pode marcar alguns linfomas B, principalmente linfoma de células do manto e leucemia linfóide crônica / linfoma linfocítico. Em linfonodos normais, tem marcação semelhante a CD3. Precisa ser analizado em conjunto com CD20 e CD3. Em ocorrendo positividade para CD20 e CD5, dirige a suspeita para os dois linfomas acima. | |
 |
|
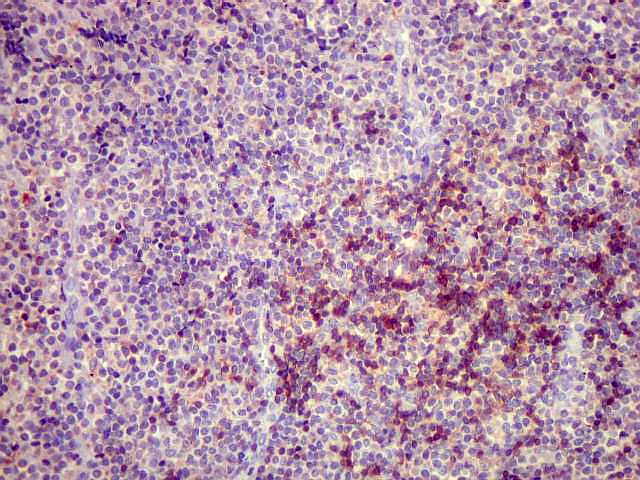 |
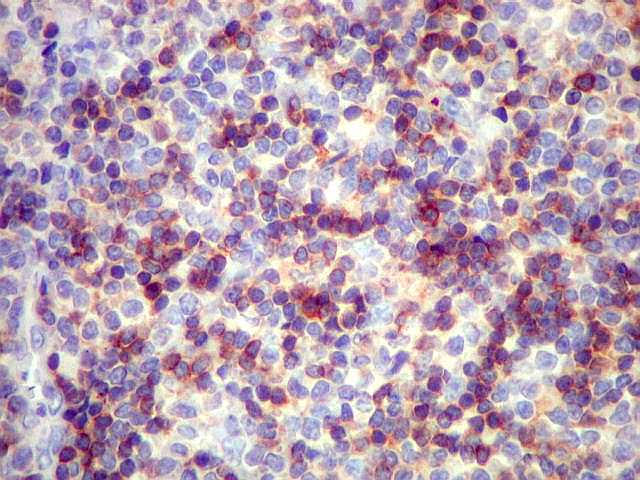 |
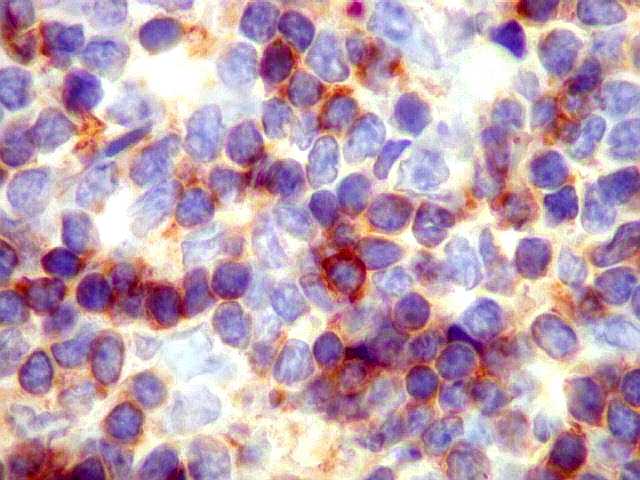 |
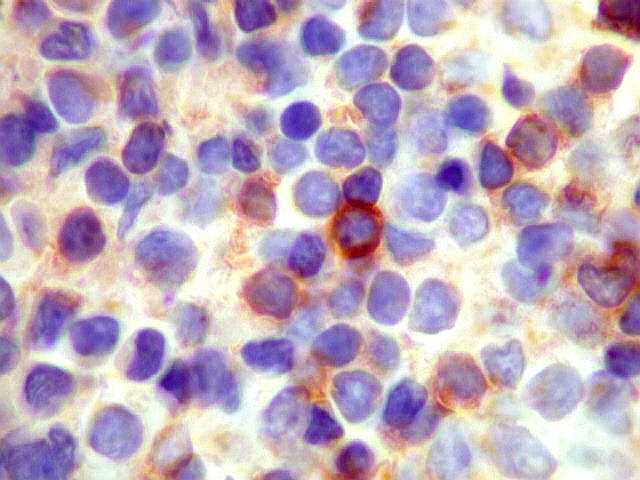 |
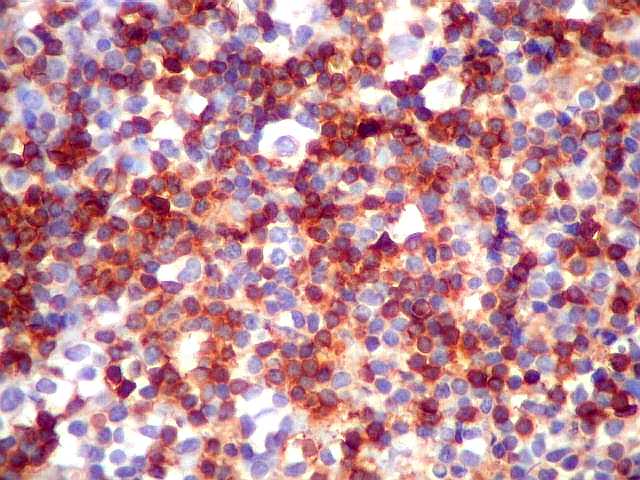
| CD57. Negativo nas células neoplásicas. É um marcador de linfomas T/NK. | |
 |
 |
 |
 |
 |
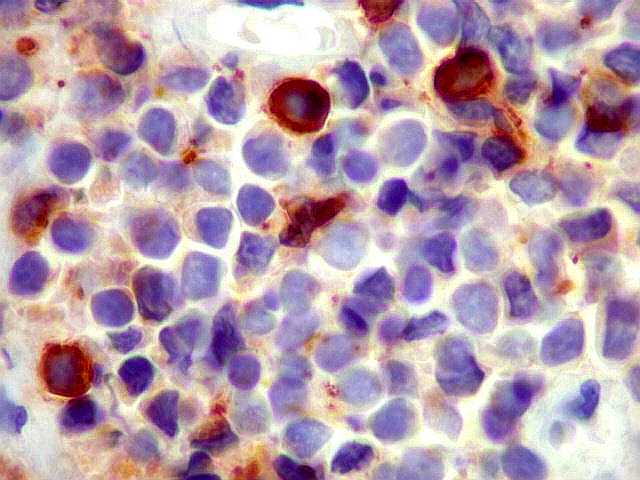 |
| BCL-1.
Também conhecido como ciclina D1, é uma proteína reguladora
do ciclo celular, identificada através da translocação
de seu gene [t(11;14)] em linfomas de células do manto. É
uma proteína nuclear detectável em cortes de parafina (a
positividade é nuclear) e é encontrada na maioria dos linfomas
de células do manto. Também pode ser expressada em leucemia
de células cabeludas ('hairy cell leukemia') e plasmocitomas,
mas com sinal mais fraco. Pode haver coloração citoplasmática
inespecífica em muitos linfomas B, que não se correlaciona
com a expressão do mRNA da proteína. Aqui, negativa,
afasta linfoma de células do manto.
Ref. Gocke CD. Immunohistology of non-Hodgkin lymphoma. in Dabbs DJ, Diagnostic Immunohistochemistry, 2nd Ed., Churchill Livingstone Elsevier, 2006, p. 140. |
|
 |
 |
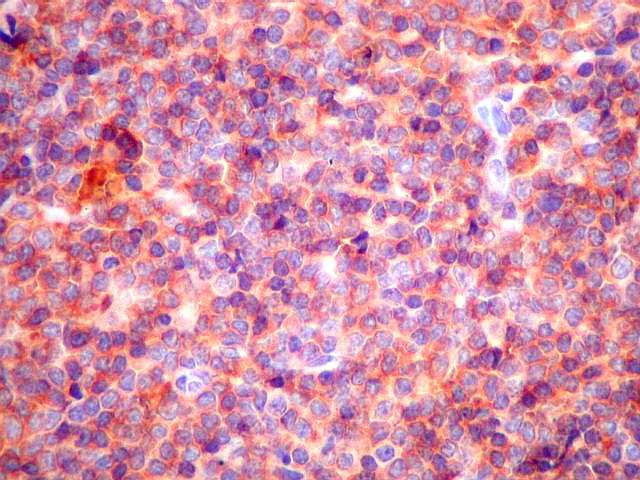
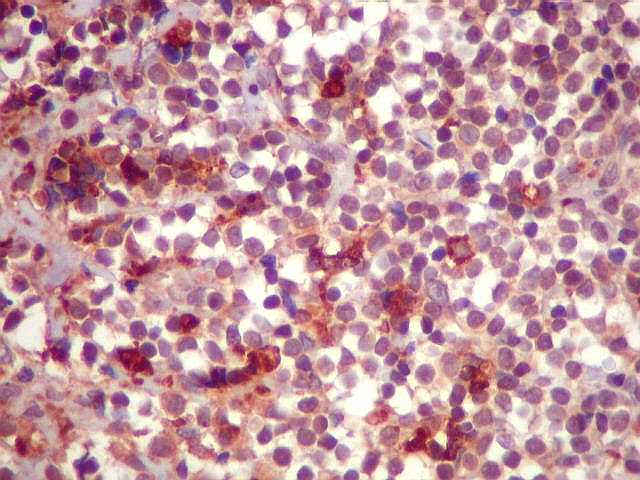
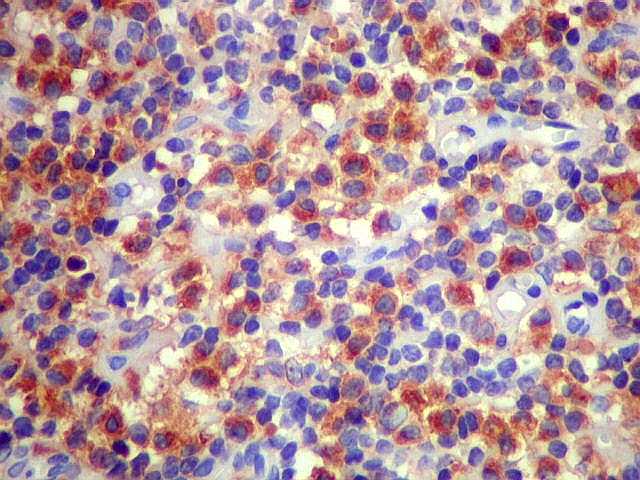
| CD68. Demonstra macrófagos esparsos entre os linfócitos tumorais. | |
 |
 |
|
 |
 |
 |
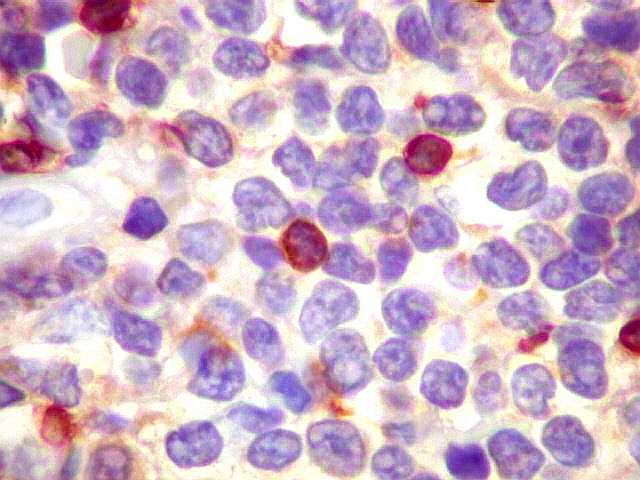
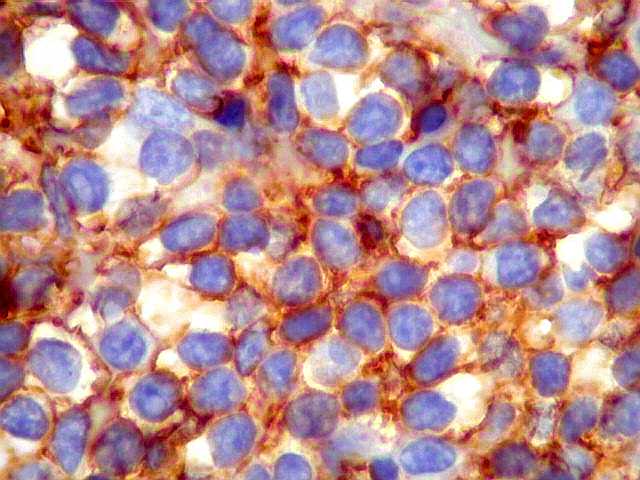
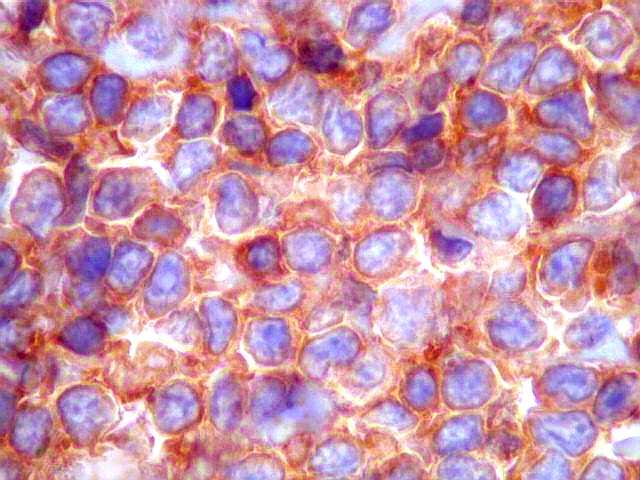
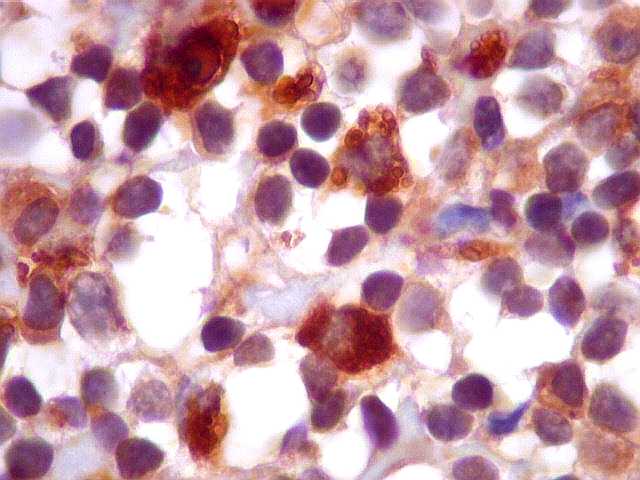
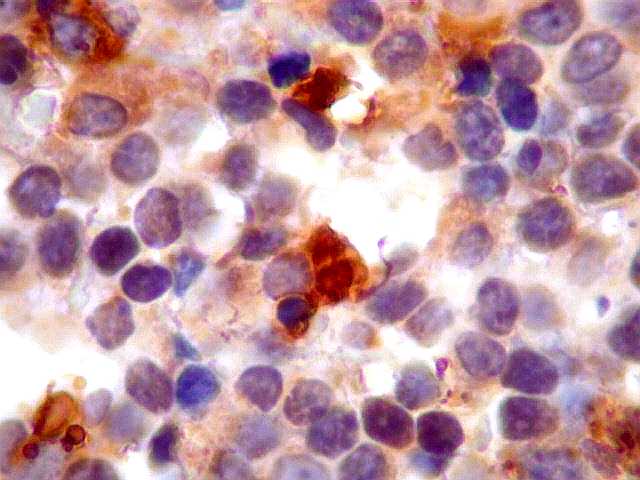
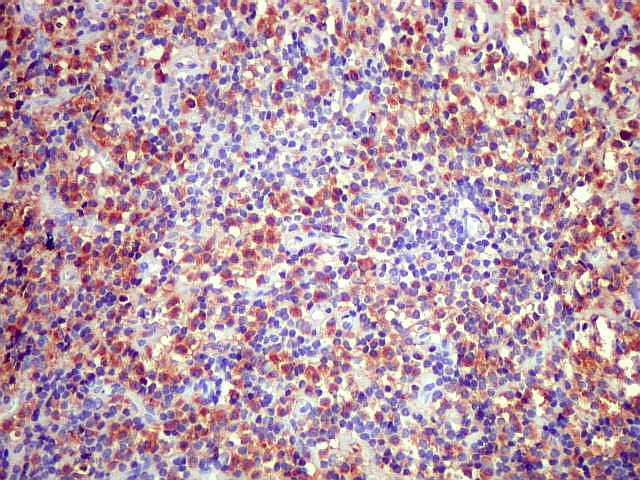
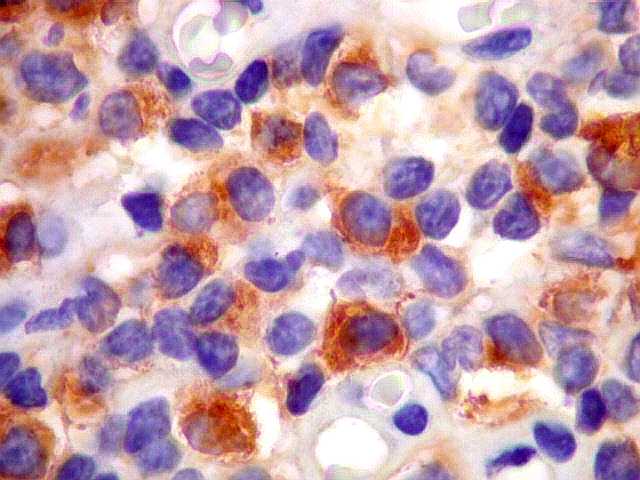
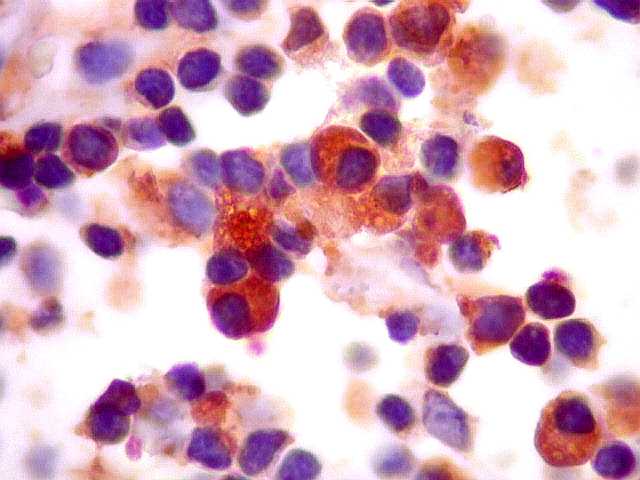
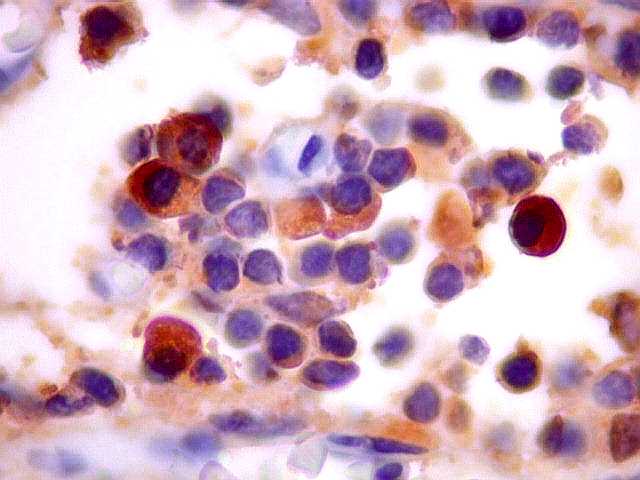
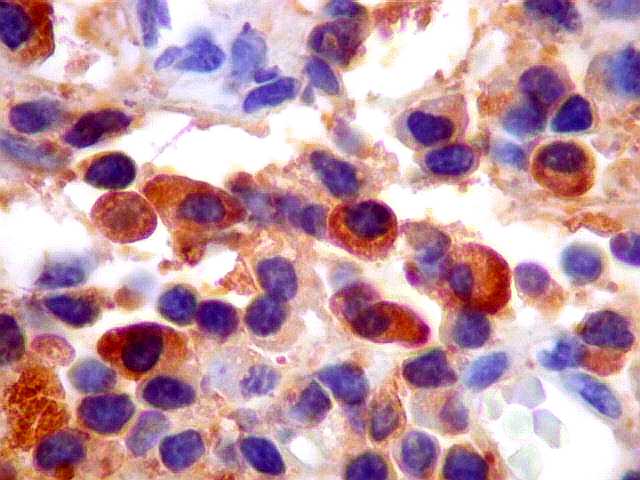
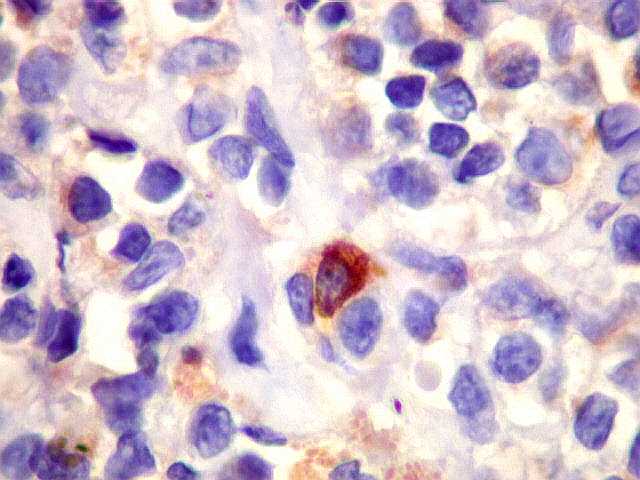
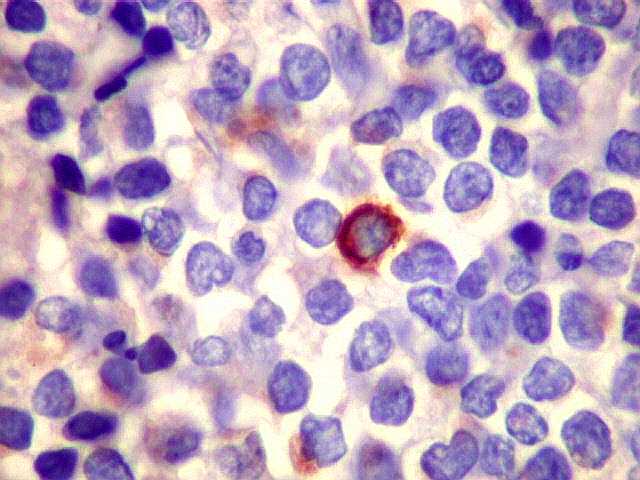
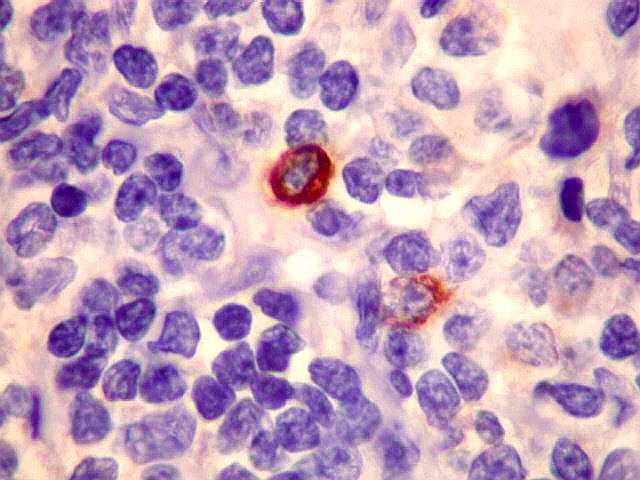
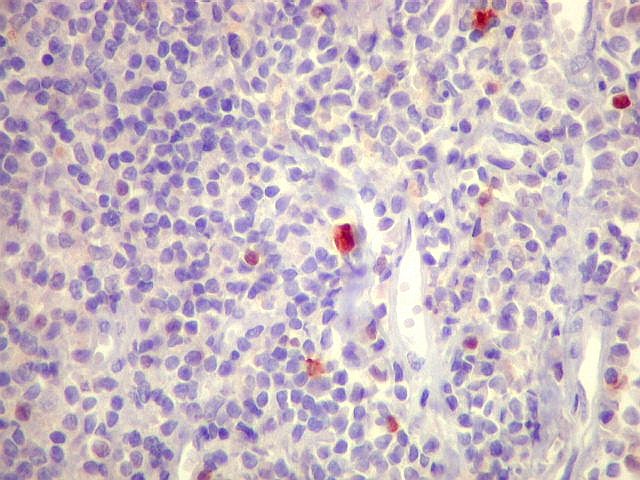
| kappa. Os plasmócitos (e algumas células linfóides sem aspecto morfológico plasmocitóide) participantes do tumor, oriundos de diferenciação dos linfócitos B neoplásicos, marcam-se para cadeia leve kappa (positividade citoplasmática), sendo negativos para cadeia lambda (quadro seguinte) (restrição de cadeias leves). | |
 |
 |
|
 |
 |
 |
 |
 |
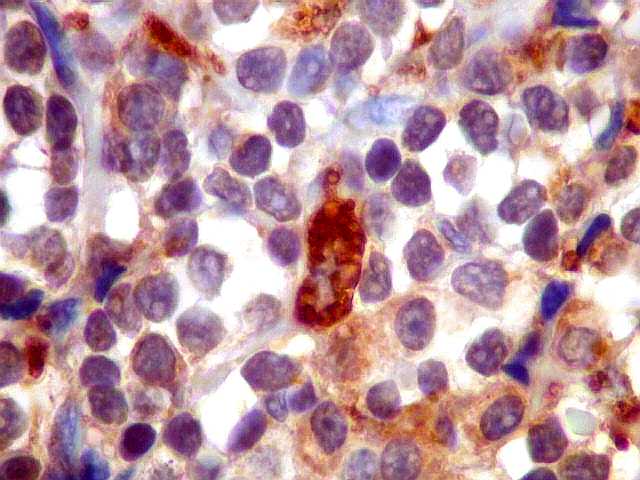
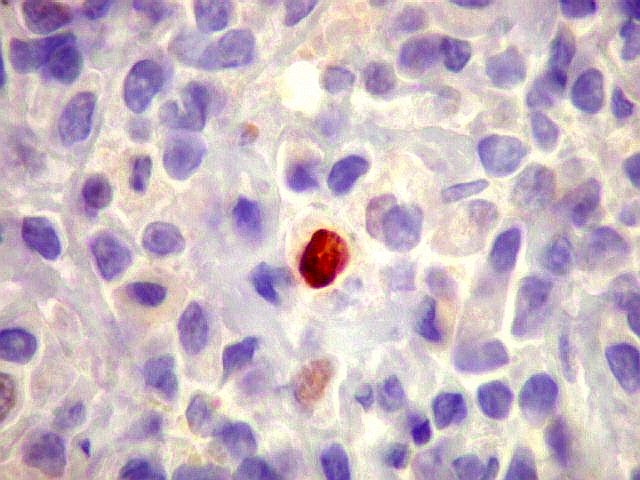
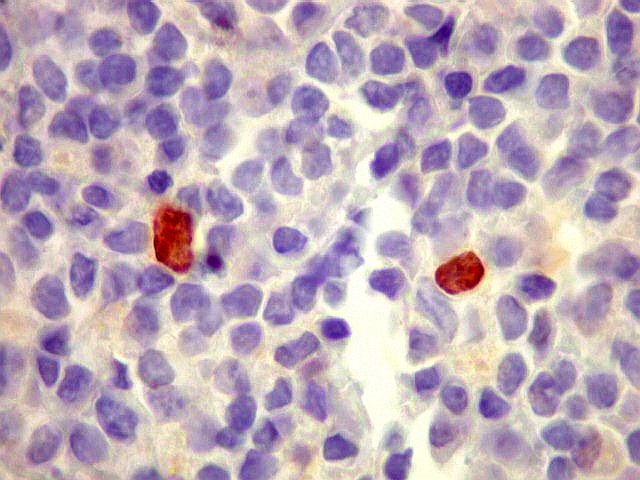
| lambda. Marcação de raros plasmócitos esparsos, considerados préexistentes ou residuais. | |
 |
|
 |
 |
 |
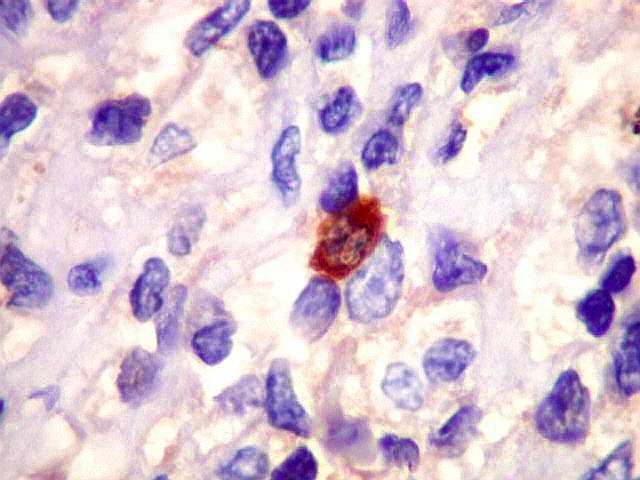 |
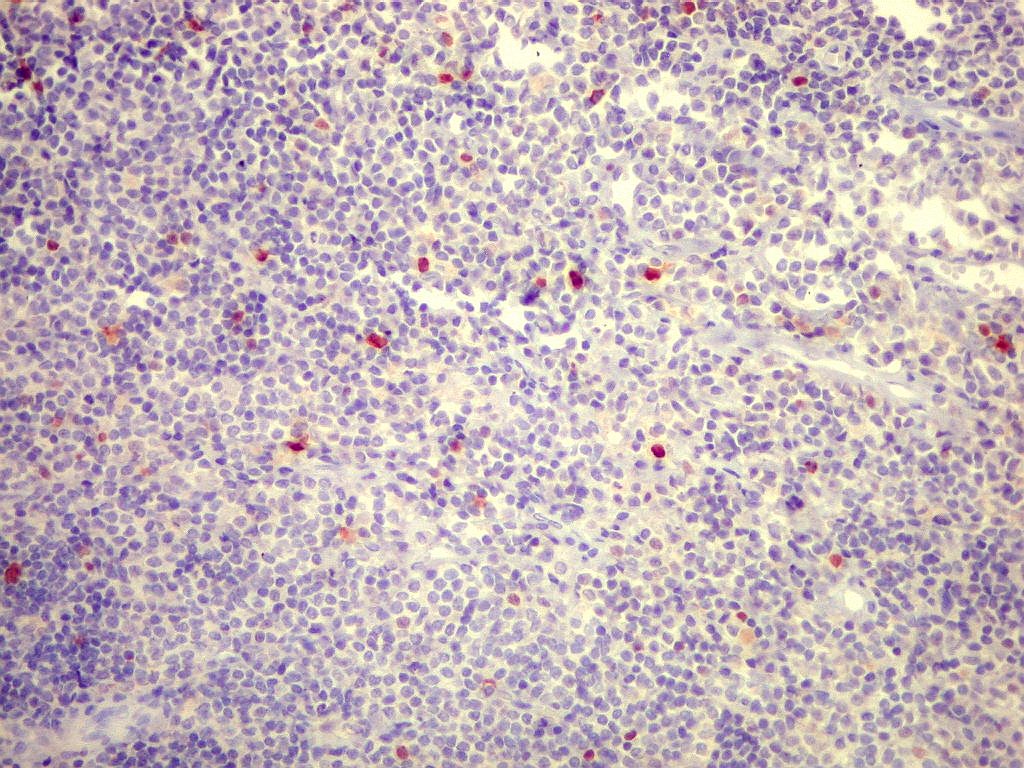
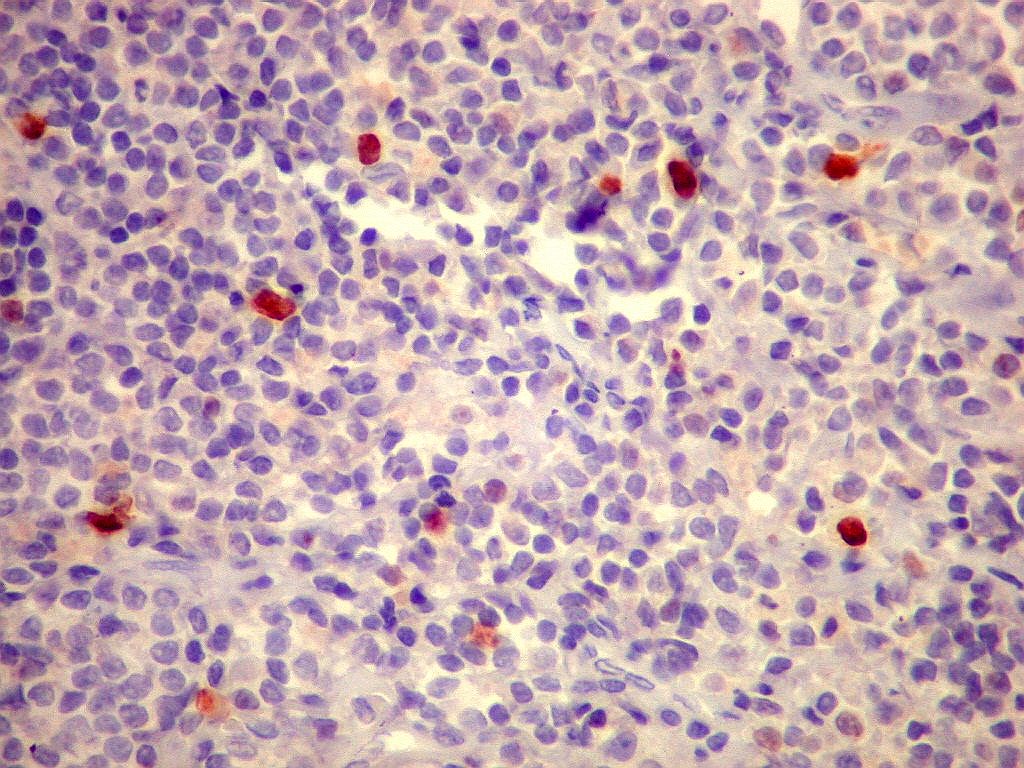
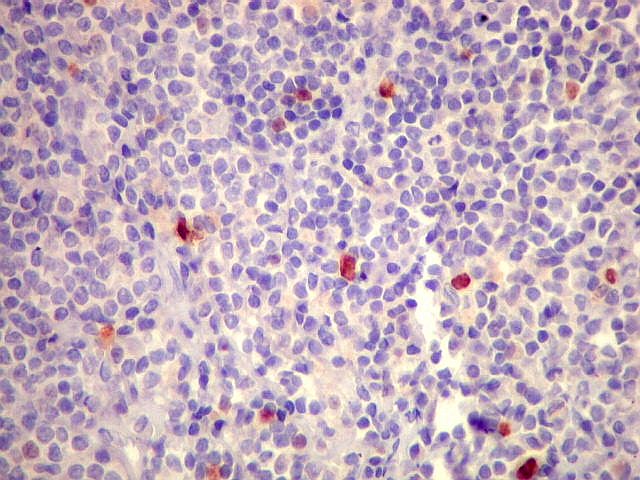
| Ki-67. Marcação de pequena proporção dos núcleos, estimada em cerca de 3%, consistente com a natureza indolente e lenta progressão do tumor. |
 |
 |
 |
 |
 |
|
 |
 |
 |
 |
 |
 |
 |
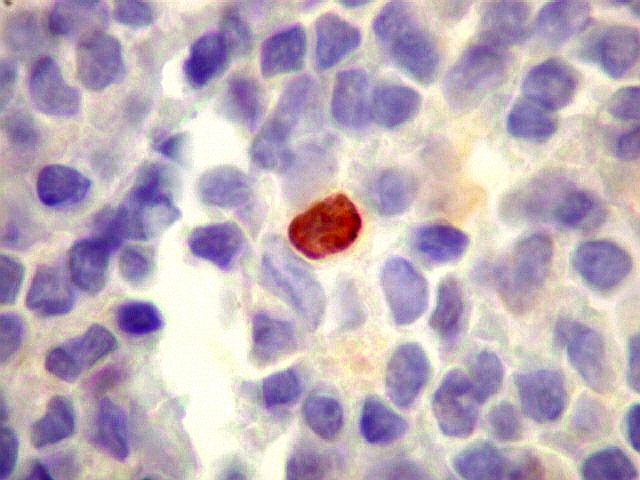 |
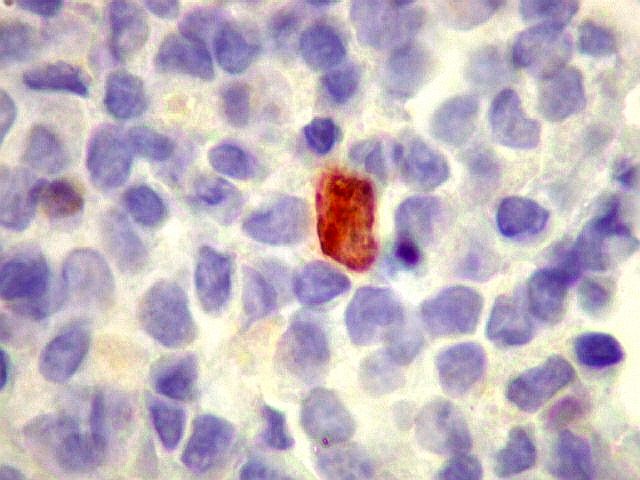
| p53. Negativo em toda a amostra. | |
 |
 |
| Diagnóstico final - linfoma não Hodgkin de imunofenótipo B, de baixo grau histológico, com diferenciação plasmocítica, mais provavelmente linfoma de zona marginal extranodal. Diagnóstico diferencial com linfoma linfoplasmocítico. |
| Comentário.Este
caso ofereceu considerável dificuldade diagnóstica
e passou por várias hipóteses até a conclusão
exposta acima. Inicialmente, na fase dos exames de imagem, a hipótese
era de meningioma da região do seio cavernoso. No ato cirúrgico,
o aspecto inusitado do tumor levou o neurocirurgião a executar apenas
uma pequena biópsia. Ao examinar os cortes de parafina, o
aspecto era de um infiltrado inflamatório crônico inespecífico
com predomínio de linfócitos e alguns plasmócitos.
Aventamos a hipótese de pseudotumor inflamatório, o que pareceu
confirmar-se com a boa resposta a corticoterapia. Posteriormente,
num reestudo de lesões problemáticas, inclusive um meningioma
rico em linfócitos e plasmócitos, a opinião do
hemopatologista foi solicitada. Este levantou a possibilidade de linfoma,
logo confirmada por estudo imunohistoquímico.
Na literatura, obtida numa pesquisa no PubMed, linfomas de dura-máter são raríssimos, e uma parte considerável corresponde justamente ao linfoma de zona marginal extranodal. Há um forte predomínio no sexo feminino.
|
| Textos.
ver abaixo, também: linfoma de células
do manto, linfoma de zona marginal, linfoma
linfoplasmocítico.
Morfologia normal do linfonodo. Linfonodos são estruturas individualizadas, circundadas por cápsula fibrosa fina, perfurada por numerosos vasos linfáticos aferentes, que se anastomosam com um seio subcapsular fenestrado. A linfa que entra neste seio vai lentamente permeando o tecido do linfonodo e coleta nos seios da camada medular, que confluem num único vaso linfático eferente no hilo. No córtex, logo abaixo do seio marginal, há vários agregados de pequenos linfócitos, os chamados folículos primários, que contêm células B imunologicamente inocentes, isto é, que nunca fizeram contato com um antígeno. A zona paracortical entre os folículos primários é preenchida por pequenos linfócitos T. Na camada medular há relativamente poucos linfócitos e número variável de plasmócitos. Quando ocorre
estimulação antigênica, após alguns dias, os
folículos primários aumentam de volume e se tornam mais claros,
constituindo os centros germinativos.
Nestes, os linfócitos B adquirem a capacidade de fazer anticorpos
altamente específicos contra o antígeno. O centro germinativo
está circundado por uma zona mais escura, a zona
do manto, que contém
principalmente células B imunologicamente inocentes. Em algumas
condições reacionais, uma outra borda de células B
com um pouco mais de citoplasma acumula por fora da zona do manto. São
chamadas células B da zona marginal
e representam uma população de células de memória
após a fase de centro germinativo. Células da zona
paracortical T também freqüentemente sofrem hiperplasia em
reações onde imunidade celular é importante, como
em infecções virais.
Constituem até cerca de 3% dos linfomas não Hodgkin nos Estados Unidos e 7 a 9% na Europa. Geralmente apresenta-se na 5ª. a 6ª. décadas, com predomínio masculino. As células neoplásicas assemelham-se às células B normais da zona do manto, que circunda os centros germinativos. Morfologia. As células tumorais podem circundar centros germinativos reacionais, produzindo uma aparência nodular em pequeno aumento, ou podem borrar difusamente a arquitetura do linfonodo. Tipicamente, as células neoplásicas constituem uma população homogênea de pequenos linfócitos, cujo núcleo varia de redondo a irregular a profundamente sulcado (cleaved). Geralmente não se observam células grandes parecendo centroblastos nem centros proliferativos, ajudando a distinguir os linfomas do manto dos linfomas foliculares e da leucemia linfóide crônica / linfoma linfocítico. Clínica. A maioria dos pacientes tem linfadenopatia quando do diagnóstico. Entre 20 e 40% tem envolvimento do sangue periférico. Sítios extranodais de acometimento pelo linfoma incluem a medula óssea, polpa branca do baço, áreas hepáticas periportais e o TGI. Há esplenomegalia em 50% dos casos. Linfomas da zona do manto não são curáveis por quimioterapia convencional. Prognóstico não é bom. Sobrevida média 3 a 4 anos. Imunofenótipo. Linfoma da zona do manto expressa CD19, CD20 (marcadores B), cadeias pesadas de imunoglobulina em padrão membrana (geralmente IgM e IgD), e cadeias leves (ou kappa ou lambda). Geralmente é positivo para CD5 e negativo para CD23, o que ajuda a distinguí-lo de leucemia linfóide crônica / linfoma linfocítico. (CD5 é expressado em células T e em um pequeno subgrupo de células B; CD23 é expressado em células B maduras ativadas). Também, linfoma do manto é caracteristicamente positivo para ciclina D1 (BCL-1). A origem deste linfoma é provavelmente numa célula B imunologicamente inocente (naïve). Genética.
O linfoma da zona do manto associa-se a uma translocação
entre os cromossomos 11 e 14, que envolve o locus das cadeias pesadas de
imunoglobulina (IgH) no cromossomo 14 com o locus da ciclina D1 no cromossomo
11. Essa translocação é detectada na grande
parte dos casos (70%) por cariotipagem ou ainda mais por hibridização
in situ examinada por microscopia de fluorescência (fluorescence
in situ hybridization, FISH). A translocação leva
à hiperexpressão do gene da ciclina D1, que promove as células
da fase G1 à fase S (de síntese de DNA), durante o ciclo
celular.
São linfomas B heterogêneos, que podem originar-se em linfonodos, baço e sítios extranodais. Inicialmente foram reconhecidos em mucosas, e por isso foram referidos como tumores do tecido linfóide associado às mucosas (mucosa associated lymphoid tissue ou MALT), portanto maltomas. Na maioria dos casos, as células tumorais predominantes lembram as células normais da zona marginal, que representam uma população de células B de memória após a fase de centro germinativo. Associação com inflamações crônicas. Os linfomas de zona marginal merecem destaque porque freqüentemente se originam em tecidos com inflamação crônica de origem infecciosa ou autoimune. Entre estes estão os tumores de glândula salivar na doença de Sjögren, na tiroidite de Hashimoto e em gastrites por Helicobacter pylori. Os linfomas de zona marginal ficam localizados por longos períodos, e só tardiamente passam por disseminação sistêmica. Podem regredir se o agente inflamatório é eliminado, como no caso da gastrite por H. pylori. Este tumor parece fazer um contínuo com a hiperplasia linfóide reacional. O processo começaria como uma reação imune policlonal. Com o tempo, algumas células adquirem mutações, levando ao aparecimento de uma neoplasia monoclonal de células B que ainda depende de células T helper para crescer. Algumas translocações, como 11:18 e 1:14, são mais ou menos específicas para linfomas de zona marginal extranodais. A partir destas mutações o tumor não responde mais a antibioticoterapia (no caso de maltomas do estômago associados a H. pylori). Com o tempo, pode haver disseminação a outros sítios e evolução a linfoma B de grandes células, o que piora consideravelmente o prognóstico. Clínica. Os linfomas B extranodais da zona marginal do tipo MALT apresentam-se em adultos, na forma de doença localizada, mais comumente no TGI, menos comumente em outros tecidos associados a mucosas, como glândula lacrimal, pulmão e mama, e ocasionalmente em locais não associados a mucosas como pele e tiróide. Na dura-máter são raríssimos (ver). Morfologia. Têm um quadro citológico heterogêneo, com abundantes células pequenas do tipo zona marginal ou semelhantes a centrocitos, células monocitóides B com citoplasma mais abundante e núcleo reniforme, e plasmócitos. As células pequenas freqüentemente infiltram o epitélio glandular para formar lesões linfoepiteliais, que podem ser ressaltadas por anticorpos para citoqueratina. Em biópsias pequenas, pode ser difícil o diagnóstico diferencial com gastrite reacional, o que pode ser auxiliado por imunohistoquímica para cadeias leves (buscando a mono- ou pluriclonalidade dos plasmócitos presentes). Imunofenótipo. Os linfomas de zona marginal são CD20 positivos (linhagem B); CD5, CD23, CD43 e BCL-1 negativos. São CD10 negativos, o que os separa dos linfomas foliculares. Como outros linfomas de pequenos linfócitos, linfomas extranodais de zona marginal são BCL-2 positivos. Fontes: Aster JC. Diseases of white blood cells, lymph nodes, spleen and thymus. In Robbins and Cotran Pathologic Basis of Disease, 7th Ed, Kumar V, Abbas AK, Fausto N (editors). Elsevier Saunders, Philadelphia, 2005, pp 682-3. Gocke CD.
Immunohistology of non-Hodgkin lymphoma. In Diagnostic Immunohistochemistry,
2nd Ed, Dabbs DJ (editor), Churchill Livingstone Elsevier, 2006, p. 147.
Definição. É uma neoplasia de linfócitos B pequenos, linfócitos plasmocitóides e plasmócitos, usualmente envolvendo a medula óssea, e, às vezes, linfonodos e baço. A distinção entre o linfoma linfoplasmocítico e outros linfomas de pequenos linfócitos B com diferenciação plasmocitóide nem sempre é nítida, especialmente com linfomas de zona do manto ou de zona marginal. Freqüentemente cursam com altos níveis séricos de uma paraproteína, usualmente do tipo IgM, secretada pelas células neoplásicas, mas esta não é necessária para o diagnóstico. O linfoma linfoplasmocítico tem semelhança superficial com a leucemia linfóide crônica / linfoma linfocítico, mas distingue-se destes porque uma fração substancial das células tumorais sofre diferenciação terminal em plasmócitos. Mais comumente, os plasmócitos neoplásicos secretam IgM monoclonal, freqüentemente em quantidades suficientes para causar uma síndrome de hiperviscosidade do sangue conhecida como macroglobulinemia de Waldenström. Ao contrário do mieloma múltiplo, a síntese de cadeias leves e pesadas é geralmente equilibrada, assim, complicações derivadas da secreção de cadeias leves livres, como amiloidose e insuficiência renal, são raras. Sinônimos.
Epidemiologia. É uma doença rara (1,5% dos linfomas nodais), que ocorre em adultos mais velhos, com idade mediana de 63 anos e discreto predomínio masculino. Locais. O tumor comumente envolve a medula óssea, linfonodos e baço. Sangue periférico também pode ser afetado. Infiltrados extranodais também podem ocorrer, inclusive pulmão, TGI e pele. Contudo, a maioria dos casos previamente diagnosticados como imunocitoma de sítios extranodais são exemplos de linfoma de zona B marginal do tipo MALT (mucosa associated lymphoid tissue) (ver). Clínica.
A maioria dos pacientes apresenta perda de peso, fraqueza e fadiga, estas
geralmente devidas à anemia por infiltração da medula
óssea. Cerca de metade têm linfadenomegalia, esplenomegalia
e hepatomegalia. A anemia pode ser exacerbada por hemólise autoimune
(cerca de 10%), causada por crioaglutininas, que são anticorpos
IgM que se ligam às hemácias a temperaturas menores que 37º
C.
Coagulopatias podem ser causadas pela ligação de paraproteínas do tipo IgM a fatores da coagulação, plaquetas ou fibrina. Contudo, paraproteínas do tipo IgM não são por si sós diagnósticas de linfoma linfoplasmocítico ou de macroglobulinemia de Waldenström, pois podem ocorrer em pacientes com outras neoplasias linfóides ou sem uma neoplasia associada. Uma minoria de pacientes inaugura o quadro com um distúrbio relacionado a IgM, como crioglobulinemia ou gamopatia monoclonal IgM, e só mais tarde desenvolvem um linfoma linfoplasmocítico. Morfologia. Medula óssea.
infiltrado nodular, difuso ou intersticial por pequenos linfócitos,
com números variáveis de plasmócitos e linfócitos
plasmocitóides (formas intermediárias entre linfócitos
e plasmócitos). A proporção entre estas células
varia e alguns tumores contêm uma população de células
maiores, com cromatina vesicular e nucléolos proeminentes. Inclusões
PAS-positivas contendo imunoglobulinas são freqüentes no citoplasma
(corpúsculos de Russell) ou no núcleo (por invaginação
da membrana nuclear, chamados corpos de Dutcher) das células plasmocitóides.
Pode haver agregados paratrabeculares e aumento reacional da população
de mastócitos.
Linfonodos e outros tecidos.
Prognóstico.
O linfoma linfoplasmocítico é uma doença progressiva
e incurável. O curso clínico é tipicamente indolente,
com sobrevida mediana de 5 a 10 anos. Como a maior parte das IgM produzidas
ficam no interior dos vasos, sintomas causados pelos altos níveis
destas macroglobulinas, como hiperviscosidade e hemólise, podem
ser aliviados por plasmaférese. Transformação
em linfoma difuso de grandes células B ocorre numa pequena proporção
de casos e tem mau prognóstico.
Fontes. Aster JC. Diseases of white blood cells, lymph nodes, spleen and thymus. In Robbins and Cotran Pathologic Basis of Disease, 7th Ed, Kumar V, Abbas AK, Fausto N (editors). Elsevier Saunders, Philadelphia, 2005. pp 681-2. Swerdlow SH et al. Lymphoplasmacytic lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th Ed, Swerdlow SH et al. (editors). International Agency for Research on Cancer, Lyon, 2008. pp 194-5. |
| Agradecimento - externamos nossos agradecimentos ao Dr. Leandro Luiz Lopes de Freitas, médico contratado na área de Hemopatologia do Depto de Anatomia Patológica, FCM-UNICAMP, que levantou a hipótese de linfoma e orientou o estudo imunohistoquímico do presente caso. |
| Para RM desta paciente, clique » | 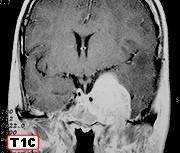 |
| Textos: linfoma de células do manto, linfoma de zona marginal, linfoma linfoplasmocítico. |
| Neuropatologia
- Graduação |
Neuropatologia -
Casos Complementares |
Neuroimagem
- Graduação |
Neuroimagem -
Casos Complementares |
Correlação
Neuropatologia - Neuroimagem |
| Índice alfabético - Neuro | Adições recentes | Banco de imagens - Neuro | Patologia - outros aparelhos | Pages in English |
|
|
|
|