|
Esfingolípides
e esfingolipidoses
(texto de apoio)
|

|
....
..
ESFINGOLÍPIDES
-
Esfingolípides
são importantes constituintes de membranas celulares.
-
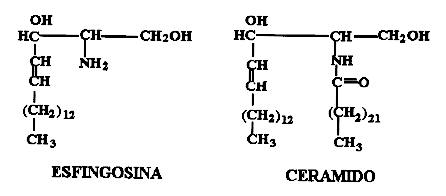
Em vez de terem
como esqueleto a molécula do glicerol, como os triglicérides,
os esfingolípides derivam do aminoálcool esfingosina.
-
Quando comparada
ao glicerol (H2COH-CHOH-CH2OH), a esfingosina tem o OH do carbono
2 substituído por uma amina (NH2)
(daí ser um amino-álcool).
-
Nos carbonos
1 e 2 há longas cadeias de ácidos graxos, que podem chegar
a 23 carbonos; muitos são insaturados. Esta molécula básica
é conhecida como ceramido.
|
..
..
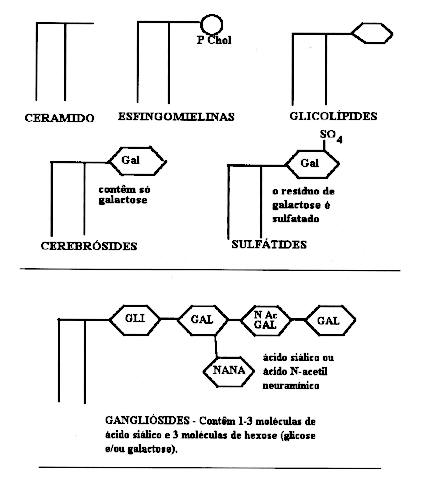
As outras
substituições são no carbono 3.
As
esfingomielinas
resultam quando no carbono 3 do ceramido há
um ácido fosfórico e colina (fosforilcolina).
Nos glicolípides,
o carbono 3 está combinado a grupos açúcar
variados e que determinam o tipo de glicolípide.
Os
cerebrósides
são glicolípides que contêm no carbono 3 uma molécula
de galactose.
Nos sulfátides,
o resíduo de galactose é sulfatado.
Nos gangliósides,
os resíduos de açúcar são múltiplos
e complexos, e incluem o ácido siálico (ou ácido
N-acetil-neuramínico).
|
..
..
| Estes lípides
complicados são constituintes essenciais de membranas, colaborando
nas suas propriedades físico-químicas. Como todos os constituintes
celulares, os esfingolípides são continuamente sintetizados
e degradados. Tanto a via de síntese como a de degradação
contêm séries de enzimas, o resultante de uma reação
sendo o substrato da próxima. Se uma das enzimas falta, a molécula
não é sintetizada, é imperfeita, ou não é
degradada. |
ESFINGOLIPIDOSES
-
São
doenças de armazenamento raras e, na maioria, autossômicas
recessivas.
-
Certas células
acumulam esfingolípides no citoplasma, devido quase sempre à
falta de uma enzima da via de degradação. Estas enzimas
são chamadas genericamente de hidrolases, porque a quebra
das ligações envolve uma molécula de água.
-
O substrato
da enzima ausente ou defeituosa acumula-se, geralmente dentro de lisossomos,
já que é nestas organelas que são encontradas as enzimas.
-
O citoplasma
fica abarrotado de lisossomos contendo o lípide não digerido.
-
Para cada enzima
que falta existe uma doença específica (às vezes mais
de uma), com acúmulo de substâncias diferentes em órgãos
diferentes, e com sintomatologia própria. Os locais mais afetados
são o cérebro, muito rico nestes compostos, e as células
do SRE. Abaixo, alguns exemplos.
|
..
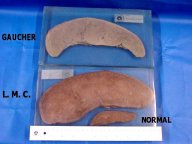
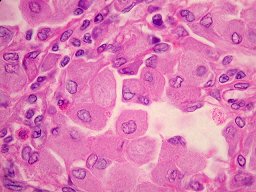
Doença
de Gaucher.
Deficiência
da enzima b
glicosidase. Acúmulo de glicosil-ceramido
(ou cerasina) em células do SRE: polpa vermelha do baço,
fígado e medula óssea. As células de Gaucher (macrófagos
muito aumentados) podem ser vistos em aspirados de medula óssea.
Há
enorme hepatoesplenomegalia. Em adultos, o sistema nervoso central é
normal. Em crianças, que têm uma forma mais grave, há
severo retardo mental.
 ......
......
..
Doença
de Niemann-Pick.
Deficiência
de esfingomielinase. Acúmulo de esfingomielina.
Há
grande hepatoesplenomegalia, retardamento mental severo e morte precoce.
..
Doença
de Tay-Sachs (Idiotia Amaurótica
Familial).
Deficiência
de hexosaminidase A. Acúmulo de gangliosídeo GM2
em neurônios de todo o SNC, inclusive retina. As células
ficam abalonadas e vão degenerando progressivamente.
Na retina,
a região da mácula, que tem menor número de corpos
celulares, destaca-se ao exame do fundo de olho como área mais vermelha
(Sinal de Tay, ou da mácula em cereja).
A doença
cursa com amaurose (cegueira) precoce e severo retardamento mental.
É
percebida já no 1o ano de vida e leva à morte em 1 a 2 anos.
Para mais
detalhes, veja 
..
..