|
|
|
|
|
|
|
|
| DOENÇA DE
CREUTZFELDT-JAKOB E OUTRAS ENCEFALOPATIAS ESPONGIFORMES.
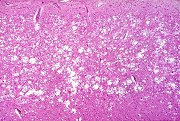
A doença de Creutzfeldt-Jakob é uma forma de demência rapidamente progressiva, que se aprofunda em questão de semanas ou poucos meses. É geralmente associada a sinais piramidais, extrapiramidais ou cerebelares, variáveis conforme o caso, mioclonias e alterações características no EEG. A doença progride inexoravelmente a coma profundo e óbito. É muito rara, causando uma morte em cada 2 milhões de habitantes por ano na Inglaterra e Estados Unidos. Desde 1957 demonstrou-se que a doença é transmissível a animais e seres humanos por inoculação intracerebral, sendo o tempo de incubação longo e muito variável (meses a vários anos). Macroscopicamente, o cérebro pode parecer pouco afetado, ou haver atrofia, com predomínio em áreas variáveis do cortex cerebral ou no cerebelo. A atrofia é mais intensa em casos com sobrevida mais longa. O exame microscópico revela a chamada encefalopatia espongiforme, caracterizada por pequenos vacúolos confluentes no neurópilo, que dão ao tecido aspecto finamente bolhoso. Além disso, há redução na população de neurônios e gliose. Característicamente, não há reação inflamatória. Em alguns casos, observam-se depósitos de amilóide no tecido, que se assemelham às placas senis da doença de Alzheimer. Em microscopia eletrônica, os vacúolos da encefalopatia espongiforme são intracelulares, situados em prolongamentos de astrócitos ou de neurônios. O agente causal da doença de Creutzfeldt-Jakob é ainda mal caracterizado. Parece tratar-se de uma partícula proteica, o príon, até hoje não havendo evidência de um componente de ácido nucleico (DNA ou RNA), como ocorre nos vírus. Não se sabe como os pacientes adquirem a doença. Esta pode ser transmitida a animais de laboratório, como macacos ou roedores, por inoculação intracerebral de amostras de cérebros. Raros pacientes são contaminados iatrogênicamente por transplantes de córnea ou preparações de hormonio de crescimento humano contaminado. Em cerca de 10% dos casos a doença é familial. O agente é altamente resistente aos métodos de assepsia habituais, inclusive formol e autoclavagem, mas é inativado por hipoclorito de sódio. Não provoca aparecimento de resposta imune no hospedeiro. Sob condições especiais, a proteína do prion purificada se condensa formando fibrilas de amilóide. Há estudos recentes indicando que a proteína do prion é codificada no DNA de neurônios humanos e no camundongo. Porém sua função normal é ainda desconhecida. Também se desconhecem que alterações tornariam a proteína patogênica. Entre as entidades relacionadas à doença de Creutzfeldt-Jakob estão o kuru, o scrapie, e a encefalopatia espongiforme bovina, conhecida como doença da vaca louca. O kuru, descrito em uma tribo na Nova Guiné, é uma forma de demência progressiva associada a ataxia e tremores (kuru, na língua indígena, significa tremor). Os nativos praticavam o canibalismo ritual, sendo a doença transmitida por este modo. O cérebro era particularmente apreciado e reservado às mulheres. A abolição do costume levou à virtual erradicação do kuru. As lesões lembram as da doença de Creutzfeldt-Jakob, com encefalopatia espongiforme predominando no cerebelo e formação de placas de amilóide no tecido nervoso. O scrapie é uma doença fatal do gado ovino e caprino, caracterizada por intenso prurido cutâneo, debilidade, incoordenação motora. O uso de carcaças de carneiros infectados por scrapie como matéria prima de ração para gado bovino teria sido causa do recente surto de encefalopatia espongiforme bovina na Grã-Bretanha. Tanto o kuru como o scrapie são transmissíveis por inoculação a animais e causam encefalopatia espongiforme semelhante à descrita na doença de Creutzfeldt-Jakob. Os agentes das três doenças (prions) são no mínimo muito semelhantes ou intimamente relacionados. |
|
..
| Módulo Neuro - Página Inicial | Outros módulos | e-mails : gradanat@fcm.unicamp.br___gradanat@unicamp.br |