|
|
|
|
|
|
|
|
|
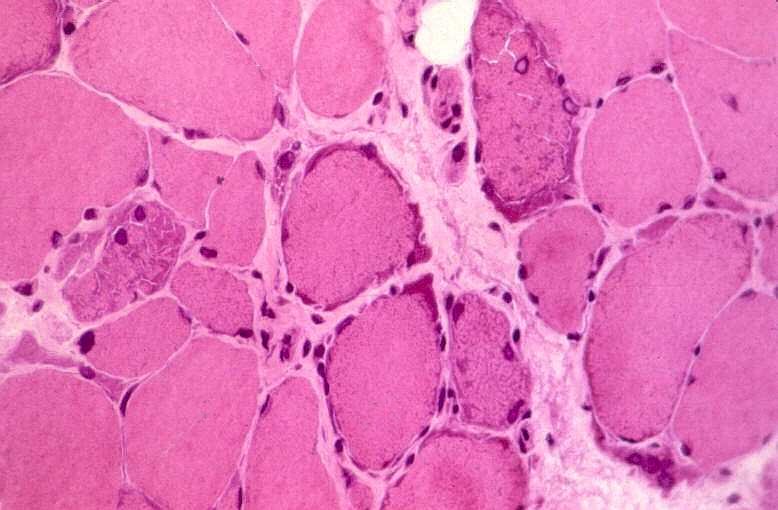
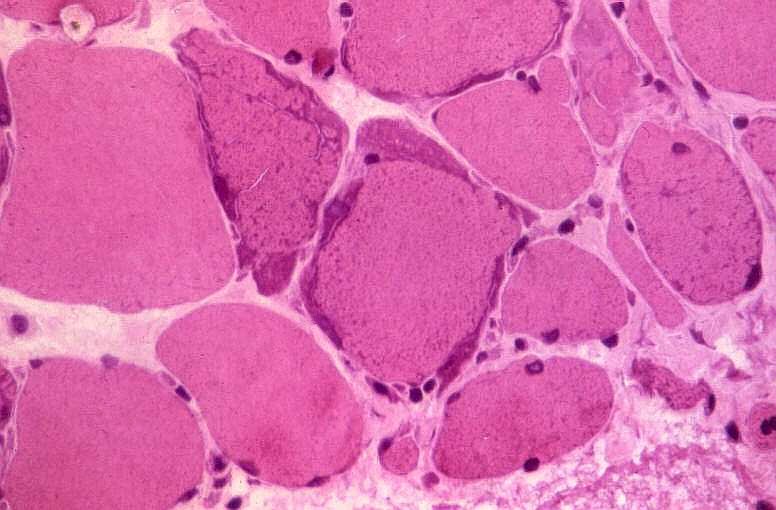
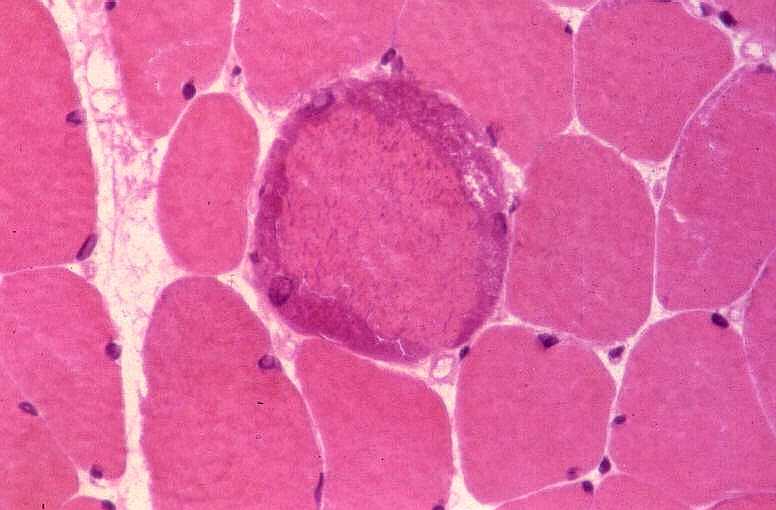
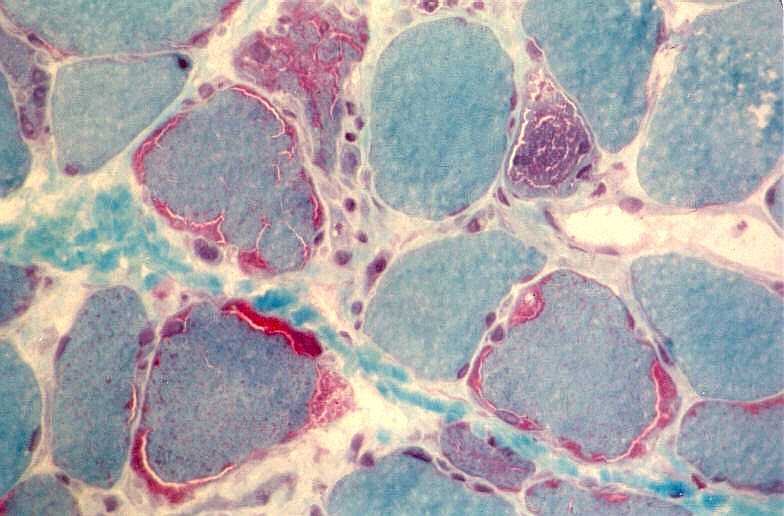
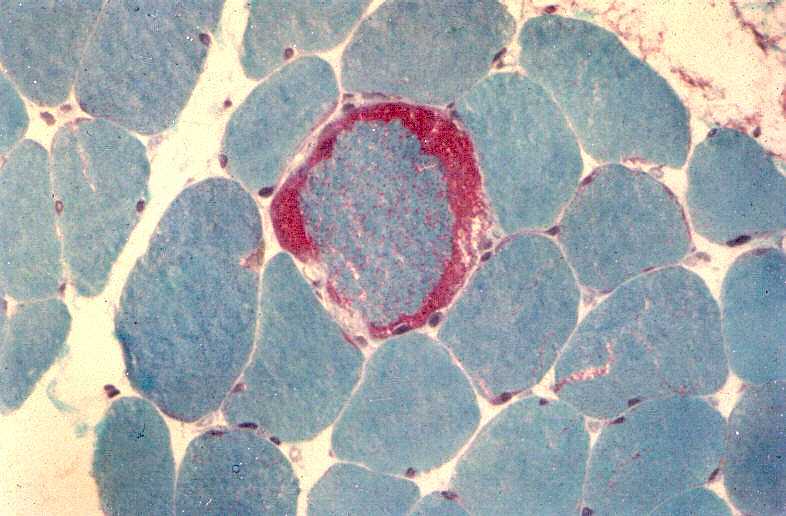
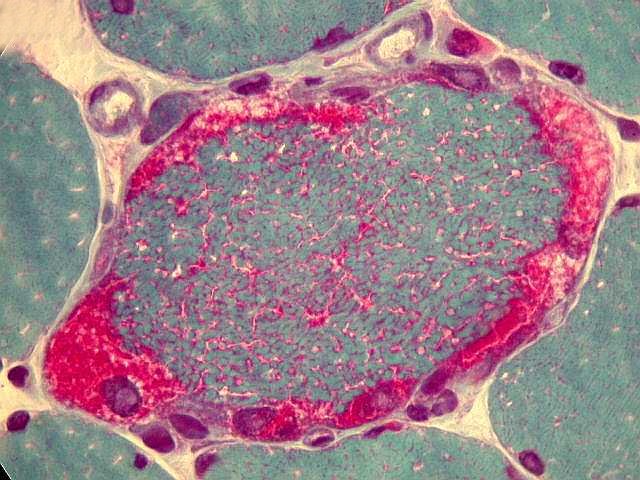
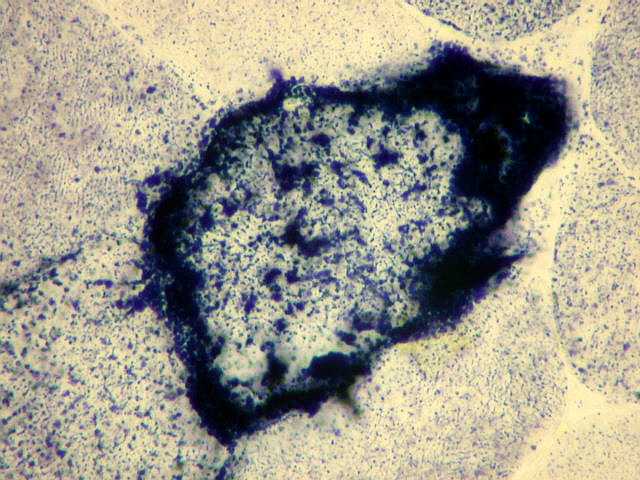
São miopatias simétricas, que acometem a musculatura proximal, reconhecidas pela primeira vez em 1966 por Shy et al. Podem ocorrer em qualquer idade, isoladamente ou em combinação com disfunção do SNC. A forma mais benigna de miopatia mitocondrial pode causar só fraqueza proximal leve, que tende a ser mais intensa nos membros superiores. Há intolerância ao exercício em quase metade dos pacientes. A progressão é extremamente lenta e o paciente pode levar vida praticamente normal. Na extremidade grave do espectro está uma miopatia infantil com fraqueza e acidose láctica que se torna evidente já na 1ª semana de vida e é fatal antes de 1 ano. A atividade de citocromooxidase é praticamente ausente no tecido muscular. Das mutações associadas à síndrome predominantemente miopática, a mais comum está na posição 3250 do genoma mitocondrial. A feição histológica que une todas as miopatias mitocondriais é a presença de ragged red fibers (ver em tricrômico de Gomori, abaixo). |
| MIOPATIA MITOCONDRIAL, HE |
 |
 |
 |
|
|
| TRICRÔMICO DE GOMORI (modificado para músculo) |
 |
 |
 |
|
|
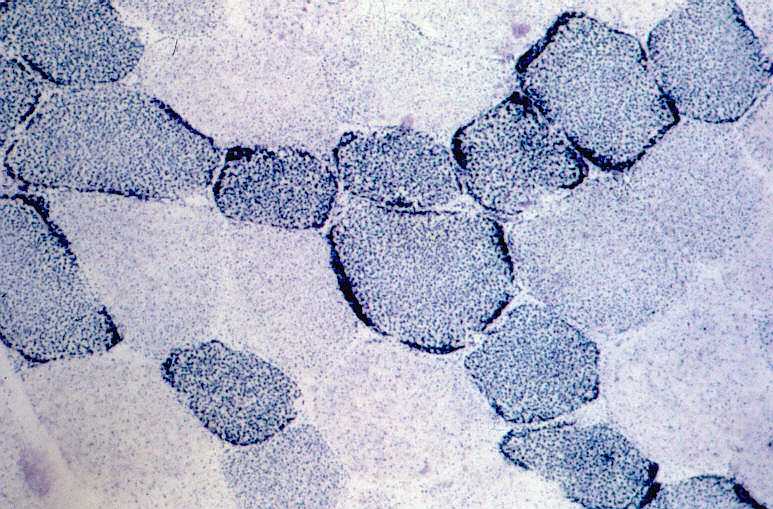
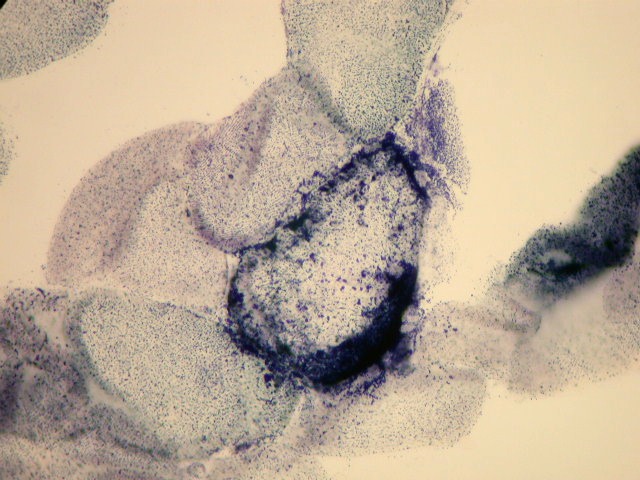
| SUCCINATO DESIDROGENASE (SDH). |
 |
 |
 |
|
|
|
|
 |
 |
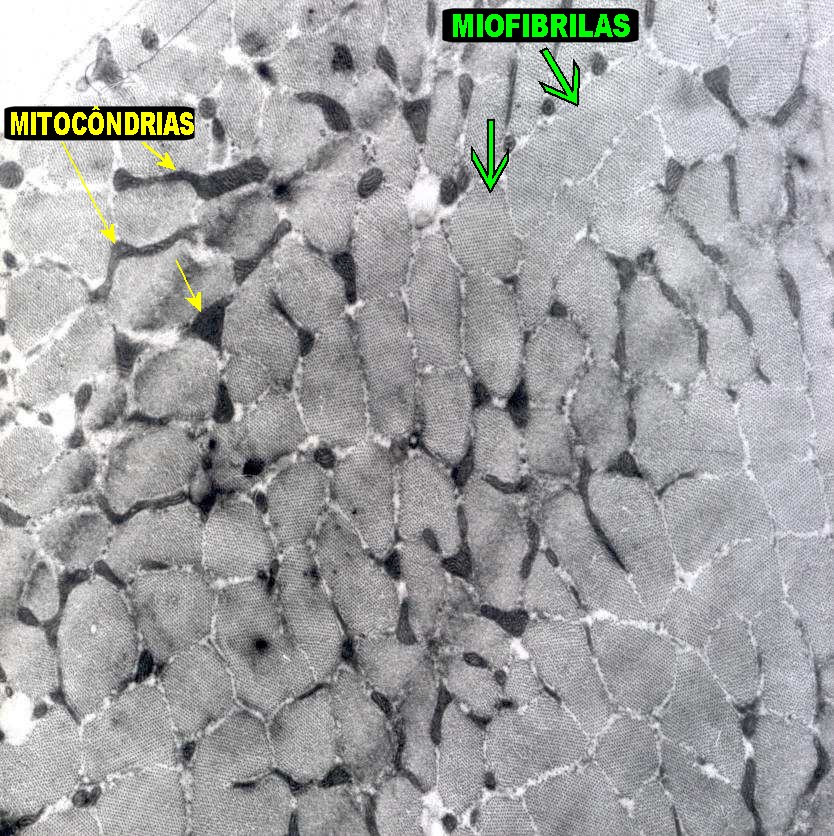
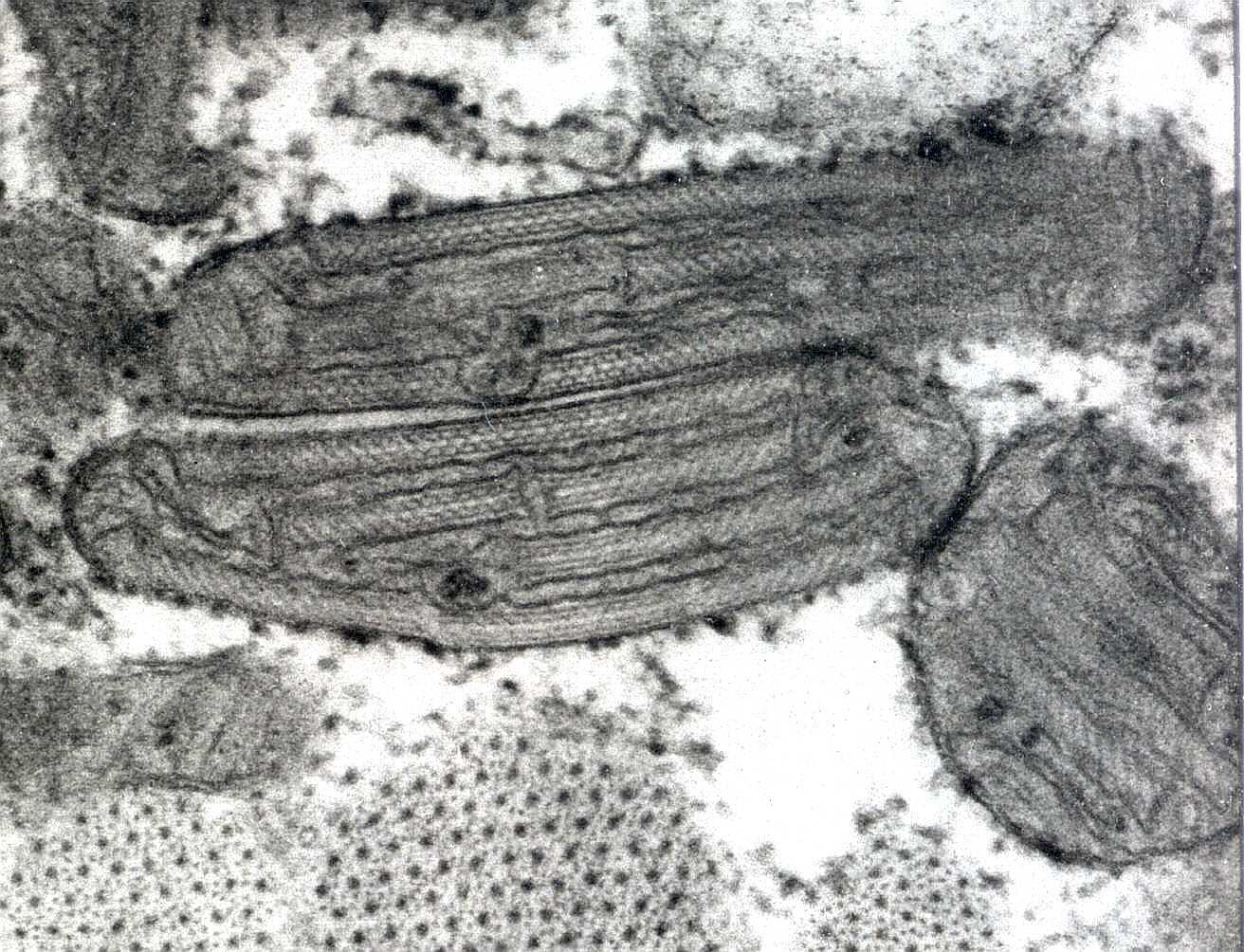
| No músculo normal, as mitocôndrias estão distribuídas de modo mais ou menos regular entre as miofibrilas, que são os feixes de actina e miosina, aqui vistos em cortes transversais de uma fibra muscular. As mitocôndrias normais têm cristas aproximadamente paralelas em meio a uma matriz eletrodensa. Há pequena variação de tamanho entre as organelas, devida em parte aos planos de corte. |
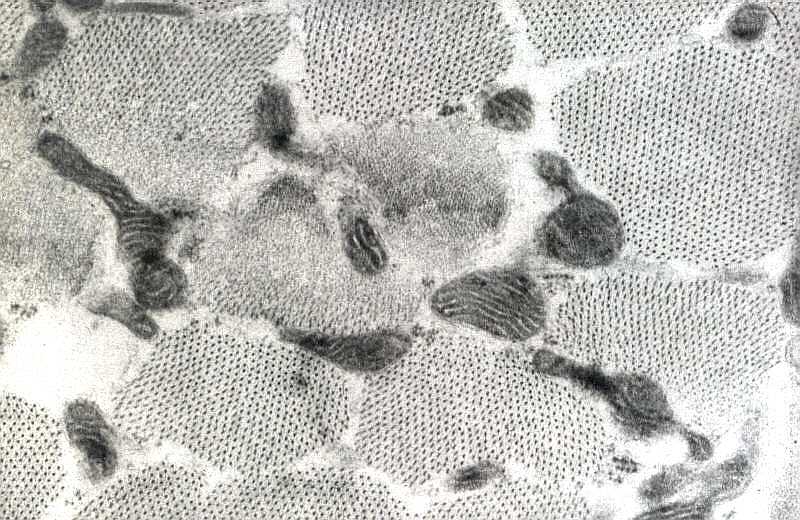
| Na miopatia
mitocondrial há aumento do tamanho
e/ou do número das mitocôndrias, mais freqüentemente
de ambos. O aumento de tamanho é designado alteração
megaconial, e o aumento de número alteração
pleoconial. Geralmente, há outras aberrações
estruturais associadas, como irregularidades na distribuição
das cristas, inclusões paracristalinas, etc.
Basicamente, as mitocôndrias aumentam de tamanho e número por serem funcionalmente ineficientes. Trata-se, de certa forma, de hipertrofia e hiperplasia vicariantes. As causas mais comuns são mutações no DNA mitocondrial, que podem ir de alterações pontuais a grandes deleções. |
 |
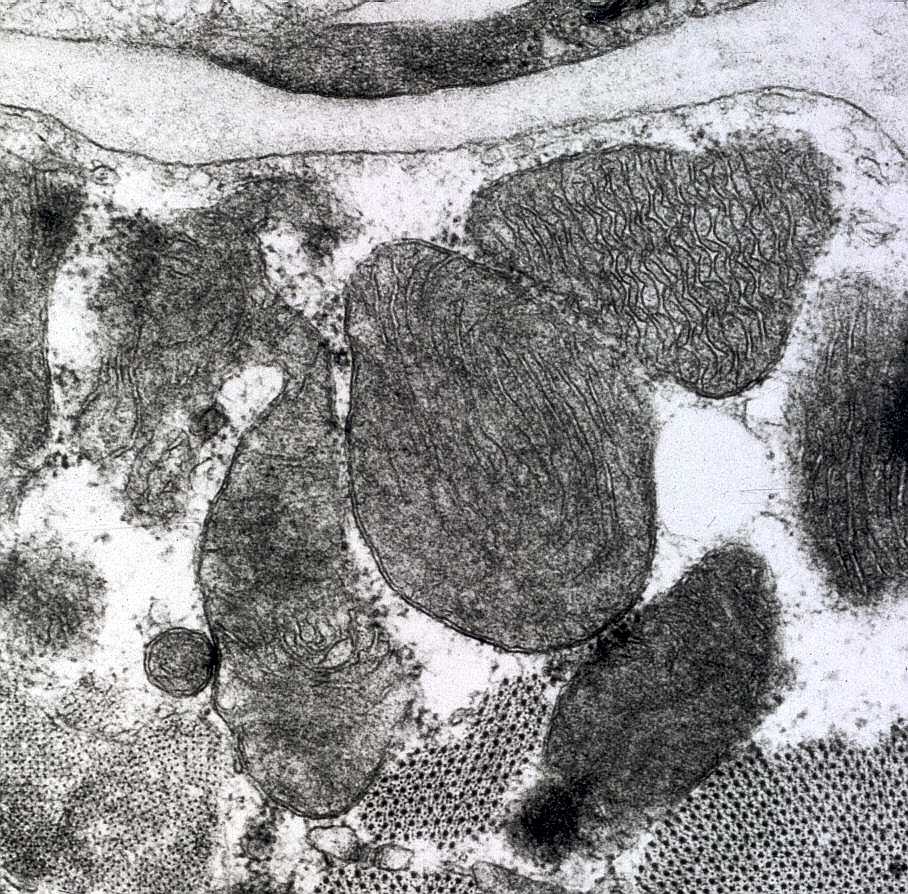 |
| Aumento numérico das mitocôndrias (alteração pleoconial) abaixo do sarcolema e anomalia morfológica: crista única paralela à superfície das organelas. | Aumento volumétrico das mitocôndrias (alteração megaconial) e aspecto ondulado das cristas. |
 |
 |
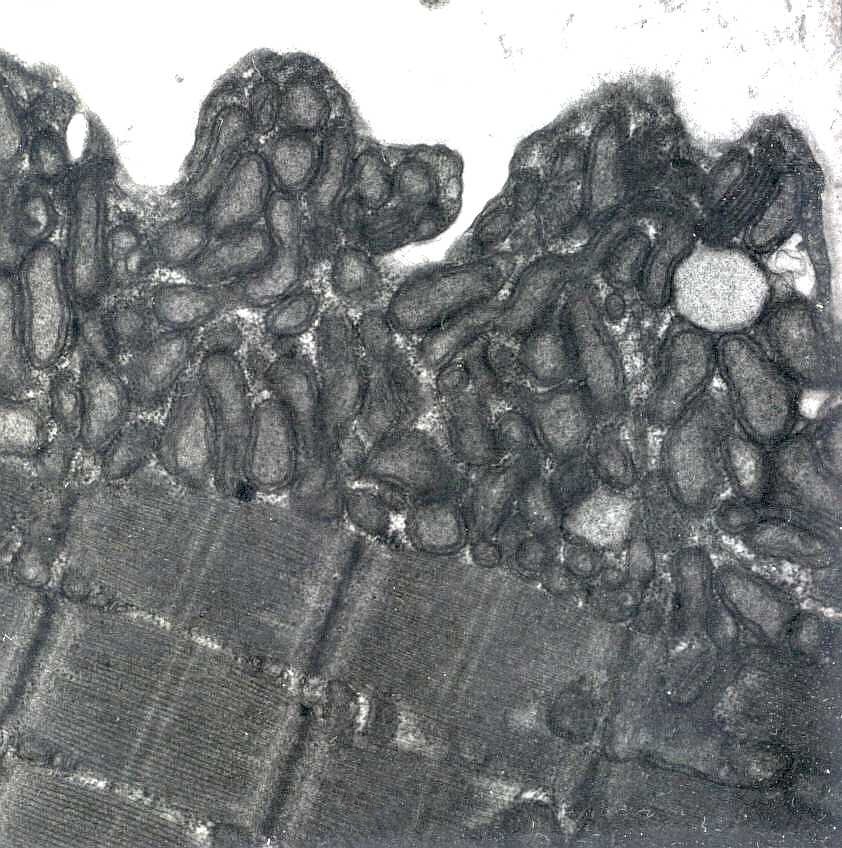
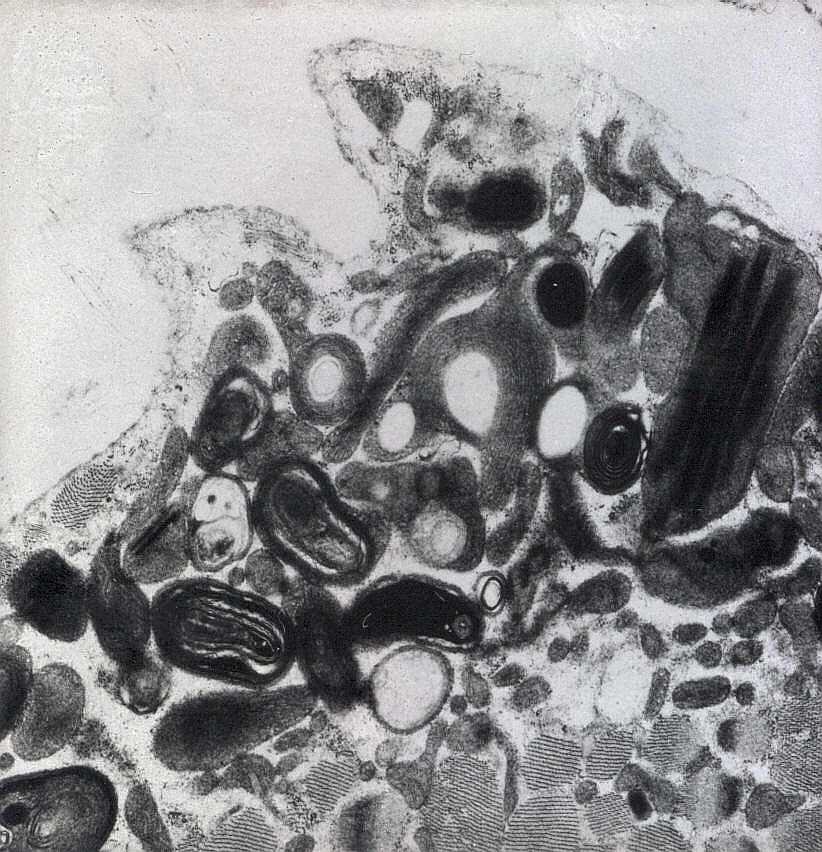
| As inclusões paracristalinas são formadas por replicação das cristas mitocondriais levando a padrões caprichosos, comparados a parede de tijolos ou pátio de estacionamento. | |
 |
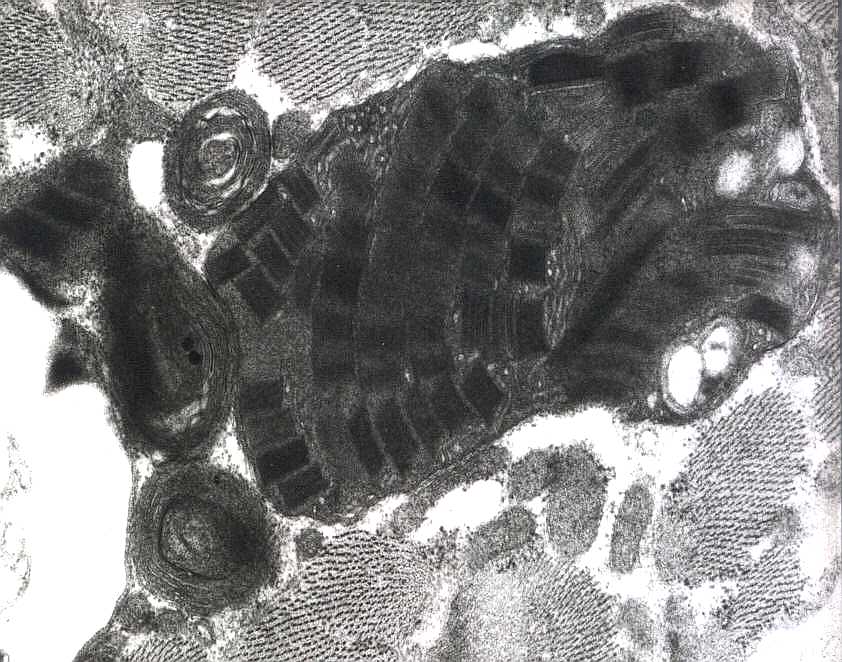 |
| Mitocôndrias aberrantes com variação de tamanho, cristas circulares, inclusões paracristalinas e mitocôndrias gigantes. A variação ultraestrutural pode ser extremamente ampla. | |
|
MITOCÔNDRIAS e SÍNDROMES MITOCONDRIAIS Mitocôndrias possuem DNA próprio. As mitocôndrias, organelas essenciais da respiração celular e síntese de ATP, possuem DNA diferente do DNA nuclear, que codifica parte das proteínas da cadeia respiratória. (A grande maioria das proteínas mitocondriais, porém, é codificada no núcleo). O mtDNA é uma pequena molécula circular de DNA presente na matriz mitocondrial na razão de 5 a 10 cópias por organela. Mutações neste DNA são comuns e de difícil reparo. São transmitidas às gerações seguintes por herança materna, diferente da herança mendeliana, já que as mitocôndrias são transmitidas ao zigoto apenas pelo óvulo, sem contribuição do espermatozóide. As mutações mais comuns são pontuais, mas pode haver duplicações e deleções de longos segmentos do mtDNA. Distribuição das mutações no mtDNA é irregular entre as células. Na multiplicação celular embrionária as mitocôndrias são repartidas entre as células filhas de forma aleatória. Se houver mutações no mtDNA, estas podem afetar só uma parte das organelas e distribuir-se de forma que certas células terão muitas mitocôndrias anormais, outras terão poucas. Se a proporção de organelas com DNA mutante exceder um valor limiar, as mitocôndrias funcionantes serão insuficientes para fornecer energia à célula. As mitocôndrias então proliferam numericamente (alteração do tipo pleoconial) e/ou aumentam de volume (alteração do tipo megaconial) incrementando a massa destas organelas no citoplasma. Podem também perder a morfologia normal (dupla membrana com membrana interna sinuosa formando cristas) e assumir configurações bizarras e formar inclusões para-cristalinas. A distribuição das mitocôndrias anômalas é casual nos vários órgãos e tecidos do corpo. Os mais afetados são aqueles de metabolismo oxidativo alto, como SNC, retina, coração, musculatura esquelética e túbulos renais. Pode haver ampla variação e combinação dos sinais e sintomas, originando as várias síndromes mitocondriais. Quadros clínicos diferentes podem ocorrer em membros da mesma família. Principais
síndromes mitocondriais
Miopatia mitocondrial. Discutida no início da página. Oftalmoplegia externa progressiva e síndrome de Kearns-Sayre. A combinação de ptose progressiva e oftalmoplegia é uma manifestação comum de doença mitocondrial. Geralmente não há diplopia ou estrabismo, ou só diplopia transitória. A oftalmoplegia externa progressiva tem relação próxima com a síndrome de Kearns-Sayre: retinite pigmentosa com início antes dos 20 anos, ataxia, bloqueio cardíaco, e aumento da proteína do líquor. Pode haver ainda surdez neurosensitiva, convulsões ou sinais piramidais. Descreve-se combinação com MELAS e MERRF que são síndromes relacionadas ao SNC (ver abaixo). Encefalomielopatia necrotisante
subaguda, ou síndrome de Leigh.
É uma doença mitocondrial familial ou esporádica com
ampla gama de manifestações clínicas. Em mais da metade
dos casos o início é no primeiro ano de vida, principalmente
nos primeiros 6 meses. O início é abrupto ou subagudo, as
vezes precipitado por episódio febril ou cirurgia.
Síndrome de neuropatia, ataxia e retinite pigmentosa (NARP). Pode incluir retardo do desenvolvimento, convulsões e fraqueza proximal. A herança é materna. É causada por uma substituição de um só nucleotídeo na posição 8993 do mtDNA. Isto causa um erro na ATP sintetase (complexo V). A severidade da doença é proporcional à quantidade de DNA aberrante no genoma mitocondrial. Epilepsia mioclônica com ragged red fibers (MERRF). Apresenta-se com epilepsia mioclônica progressiva ou ataxia mioclônica. Mioclonias em uma criança ou adulto jovem são a feição mais típica. Há vários tipos de crises convulsivas que podem estar associadas, e várias são desencadeadas por luz forte. A ataxia tende a piorar progressivamente, eclipsando as mioclonias e convulsões, mas pode permanecer relativamente leve. A miopatia é geralmente inaparente. A observação de anormalidades mitocondriais nas fibras musculares é necessária para o diagnóstico clínico. Pode ainda haver surdez, declínio mental, atrofia óptica, oftalmoplegia, lipomas cervicais, baixa estatura ou neuropatia. A maioria dos casos é familial ou mostra herança materna. Quanto mais precoce o início, mais grave a doença. Quando o início é na 1ª década, geralmente o óbito se dá antes da 3ª. Há proporcionalidade entre a quantidade de mtDNA anormal e a severidade. 80% dos pacientes com MERRF têm mutação pontual em mtDNA 8344, que codifica um RNA de transferência. Miopatia mitocondrial, encefalopatia, acidose láctica e episódios tipo AVC (MELAS). O desenvolvimento é inicialmente normal, seguido por retardo de crescimento, convulsões generalizadas ou focais e episódios que parecem AVC isquêmico ou acidente isquêmico transitório. Os episódios isquêmicos geralmente regridem, mas podem levar a encefalopatia progressiva. Pode haver acidose láctica. O CT pode mostrar várias regiões de baixa densidade na substância branca sem correlação com queixas ou achados clínicos A maioria dos pacientes tem fibras do tipo ragged red mas fraqueza ou intolerância ao exercício são raras. Cerca de 80% dos pacientes com MELAS têm mutação pontual no sítio 3243 ou num locus alternativo. Ambos codificam um RNA de transferência. Herança materna é comum, mas há casos esporádicos. Cerca de metade dos casos de mutação pontual no sítio 3243 estão associadas a MELAS. Admite-se que anormalidades no genoma mitocondrial das células endoteliais de vasos cerebrais seria a base para os episódios isquêmicos e enxaquecas. Diagnóstico
de doenças mitocondriais.
Na investigação familial, procurar história de doenças na infância, inclusive morte no período neonatal, e de convulsões. Atualmente há testes comerciais para as mutações pontuais mais comuns (3243 e 8344). Dosagem de lactato e piruvato no sangue em repouso e após exercício. Biópsia muscular ragged red fibers, aspecto do músculo no SDH, COX (citocromo-oxdase) e microscopia eletrônica. Na síndrome de Leigh e em MELAS TC e RM podem mostrar lesões cerebrais. O diagnóstico final é clínico e baseado nos antecedentes familiares, testes laboratoriais, e biópsia muscular. Sinais e sintomas isolados como demência, fraqueza muscular, epilepsia, surdez neural, enxaqueca com AVCs , epilepsia mioclônica, cardiomiopatia e baixa estatura devem levantar hipótese de doença mitocondrial no diagnóstico diferencial. Refs.
|
..
| Módulo Neuro - Página Inicial | Outros módulos | e-mails : gradanat@fcm.unicamp.br___gradanat@unicamp.br |