| Astrocitoma
/ ganglioglioma desmoplásico infantil (DIA/DIG).
Definição.
Grandes tumores cerebrais císticos na primeira infância, que
envolvem o córtex cerebral e leptomeninges, e são freqüentemente
aderidos à dura-máter. O prognóstico é geralmente
bom após cirurgia. Histologicamente, são compostos por astrócitos
neoplásicos associados ou não a uma população
variável de neurônios, em meio a proeminente estroma desmoplásico.
Correspondem a grau I da OMS.
Histórico.
O astrocitoma desmoplásico infantil foi individualizado em 1982
por Taratuto et al. como astrocitoma meningocerebral aderido à dura,
com reação desmoplásica. Em 1987, VandenBerg et al.
descreveram tumores neuroepiteliais supratentoriais desmoplásicos
da infância com diferenciação divergente (ganglioglioma
desmoplásico infantil) com quadro clínico semelhante.
Em ambos tumores havia também agregados de células neuroepiteliais
imaturas.
Incidência.
São raros. Em uma série de 6500 tumores do sistema nervoso
em todas idades foram relatados 22 casos (0,3 %). Em outra série,
de tumores cerebrais da infância, corresponderam a 1,25%. Há
cerca de 80 casos descritos. A incidência etária varia de
1 a 24 meses, com preferência pelo sexo masculino de 1,5:1.
Há, porém, casos relatados até os 25 anos.
Localização.
São sempre supratentoriais, envolvendo mais de um lobo, preferencialmente
o frontal e parietal, seguidos pelo temporal e occipital.
Clínica.
Quadro de curta duração, com aumento do perímetro
cefálico, alargamento e tensão das fontanelas, letargia e
sinal do sol poente. Pode haver convulsões, hipertonia, sinais de
localização e paralisias de nervos cranianos.
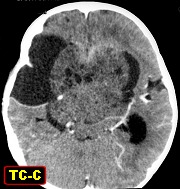
Imagem.
Na TC, aparecem como grandes massas hipodensas, císticas, com um
componente sólido isodenso ou hiperdenso. Este é superficial,
estende-se sobre as meninges em forma de placa e sofre impregnação
por contraste. Geralmente, a porção sólida é
superficial e a cística é profunda. Em RM, a porção
sólida é isointensa em T1 e heterogênea em T2, com
captação de contraste. Edema é pouco proeminente e
pode faltar. O tamanho dos tumores quando da descoberta chega a ser alarmante.
O grande componente cístico é o maior responsável
pelo efeito de massa. É raro haver comunicação com
o sistema ventricular. Pode haver erosão da tábua óssea
interna.
Macro.
Tumores são grandes, chegando a 13 cm. Há cistos uni- ou
multiloculados preenchidos por líquido claro ou xantocrômico.
A porção sólida superficial é primariamente
extracerebral, envolvendo córtex, leptomeninges, aderida à
dura, com cor esbranquiçada ou acinzentada e consistência
firme, devida à riqueza em fibras do tecido conjuntivo.
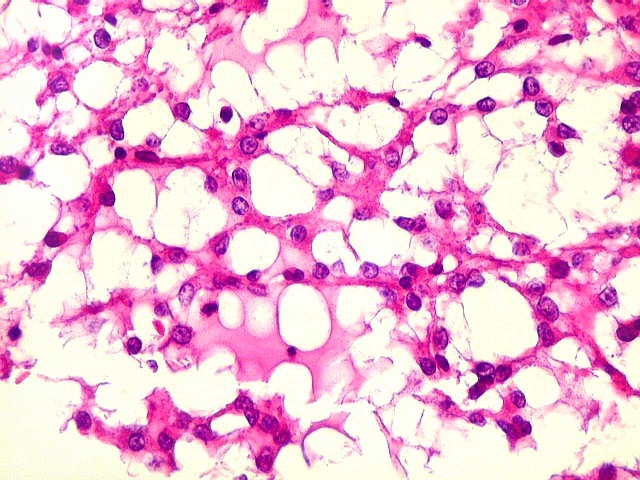
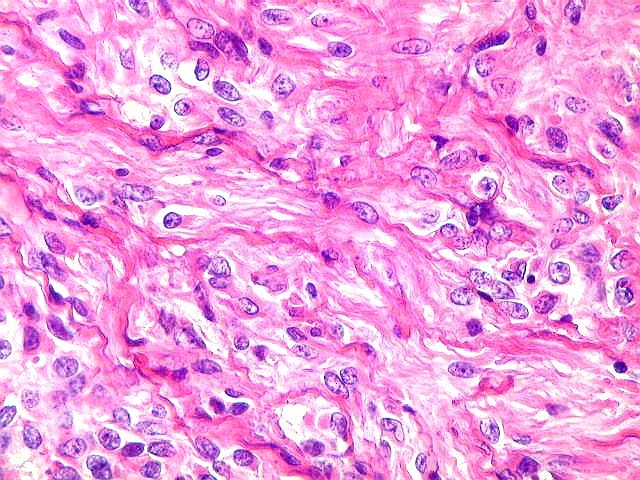
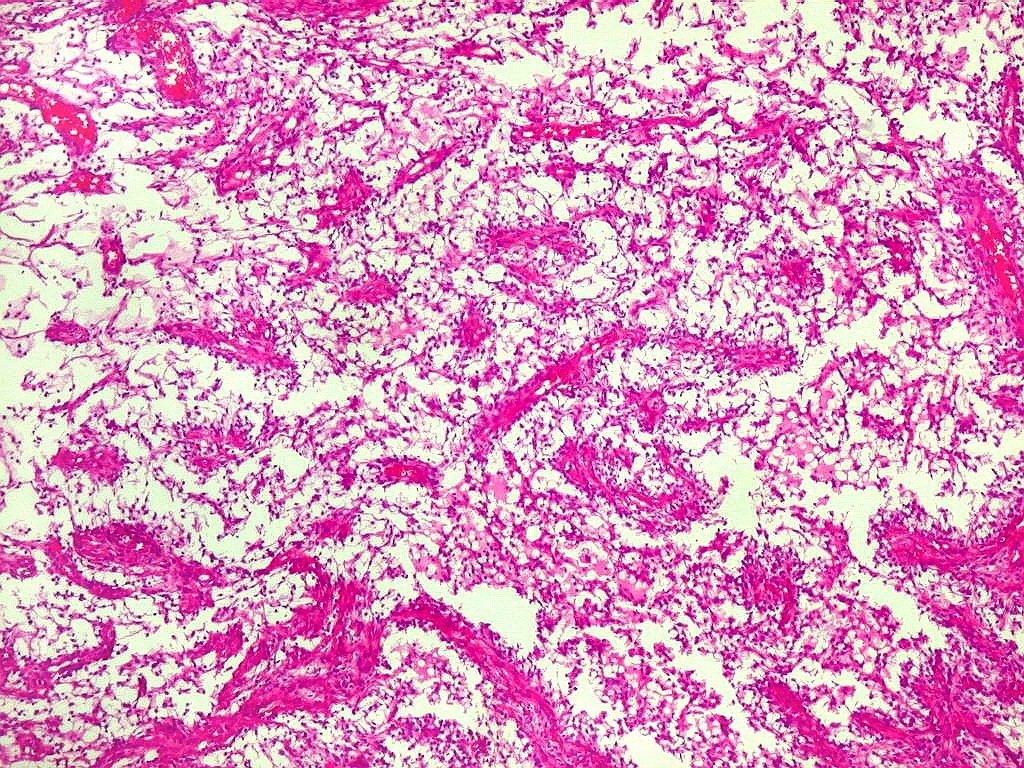
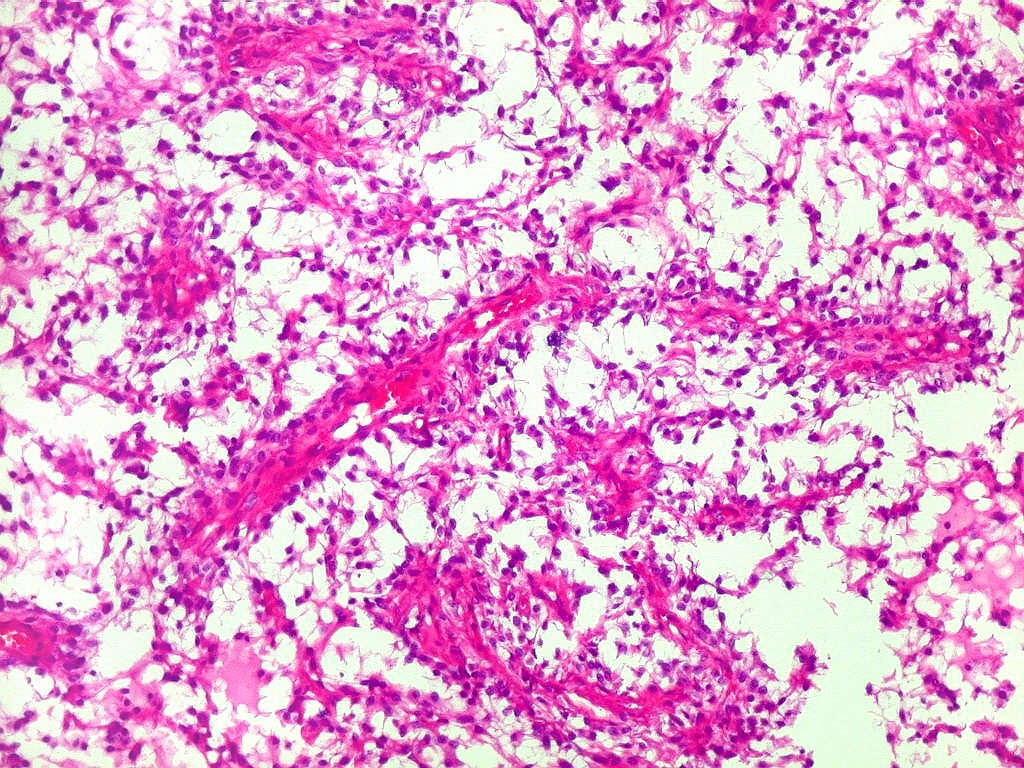
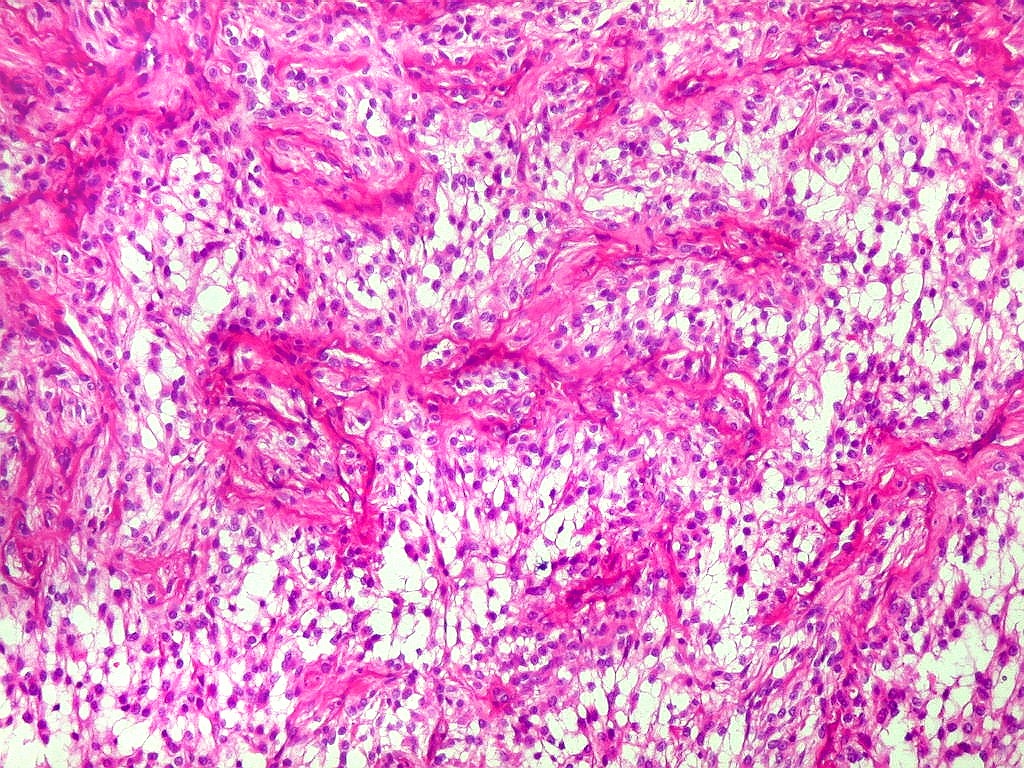
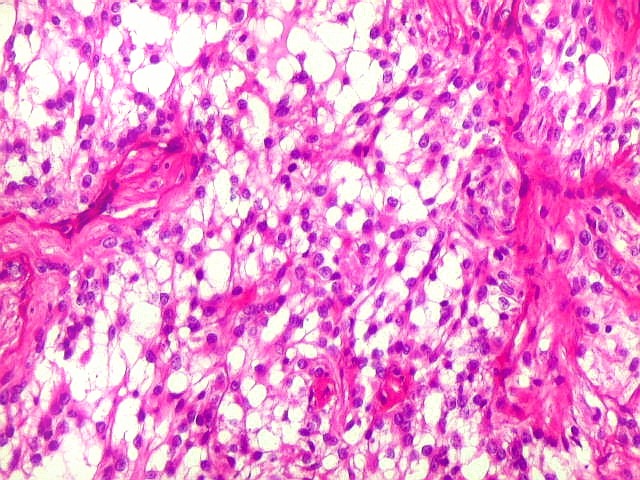
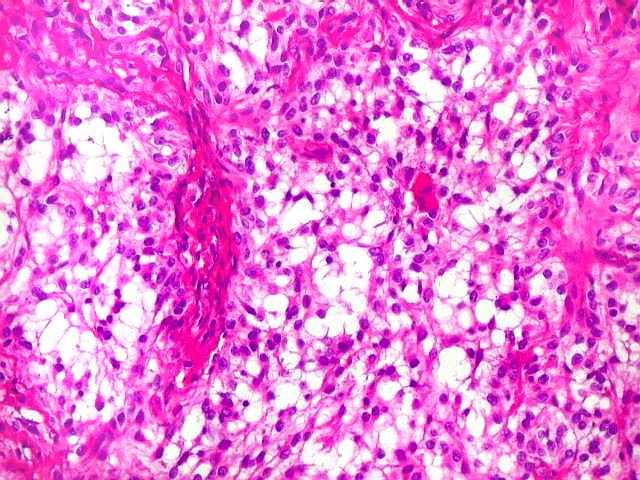
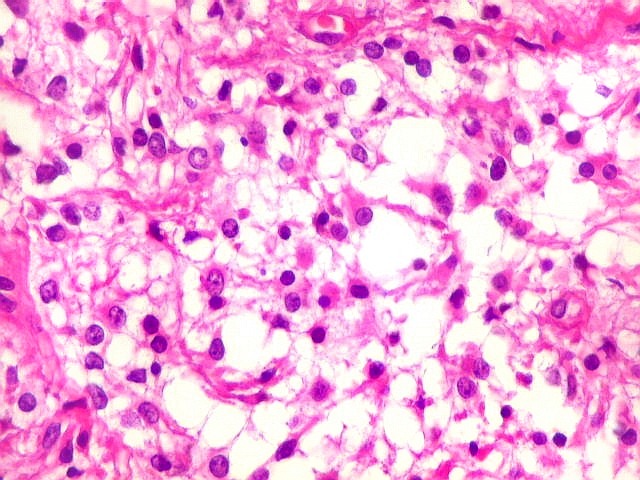
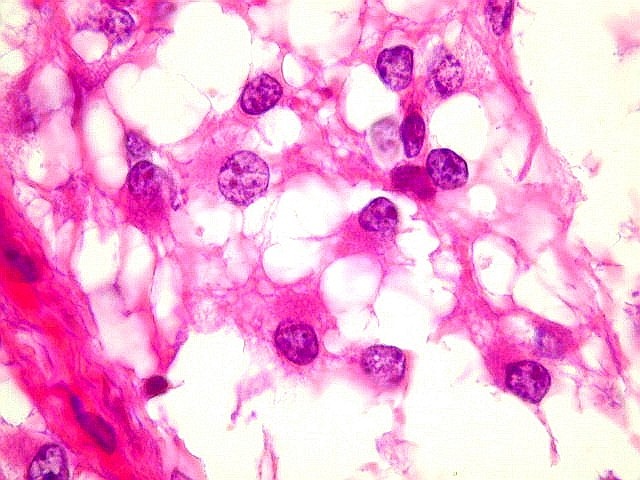
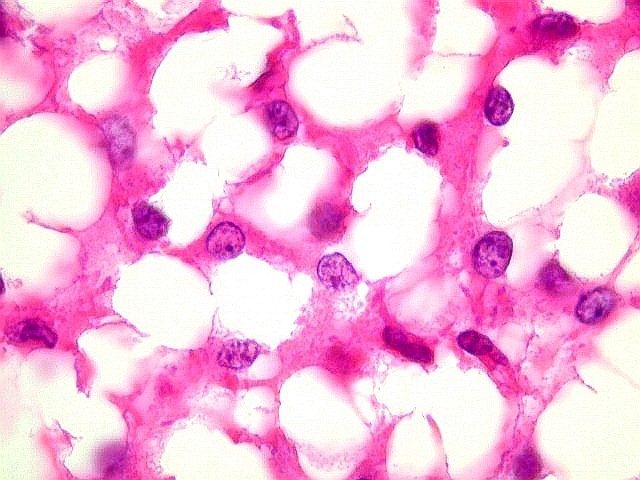
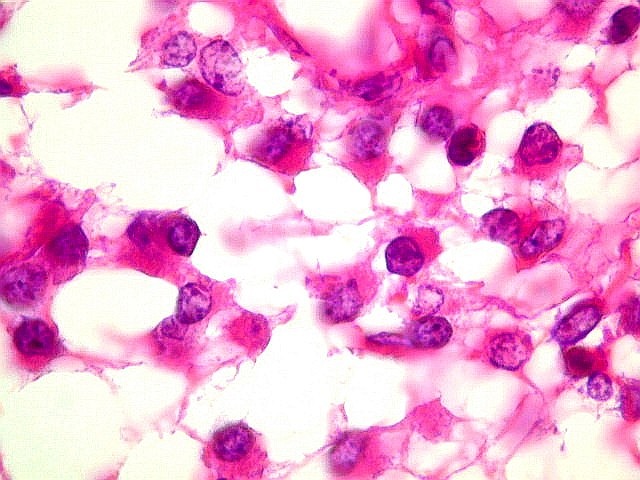
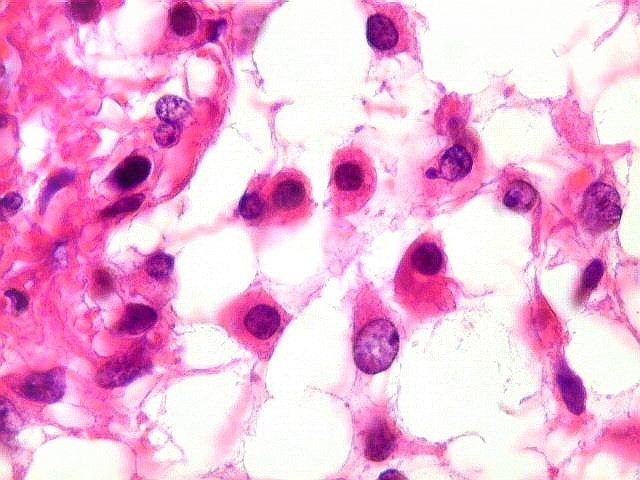
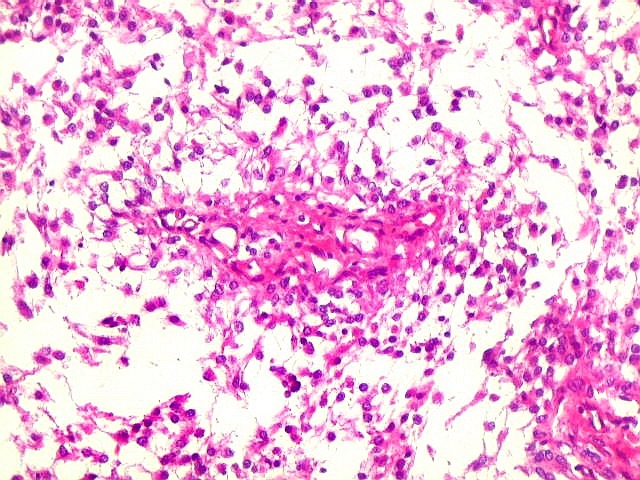
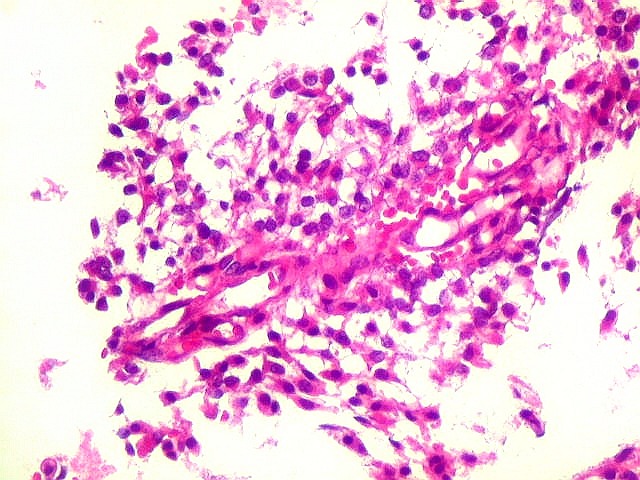
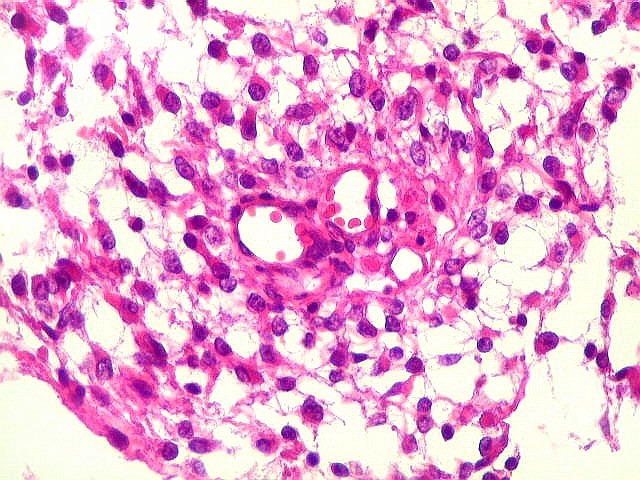
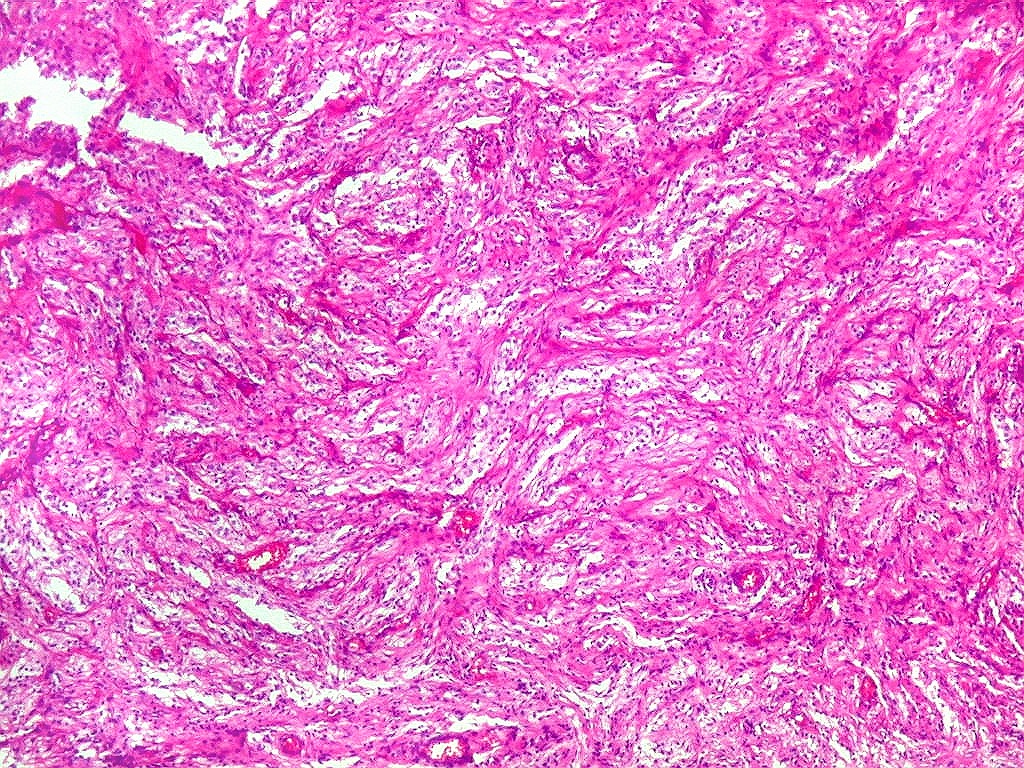
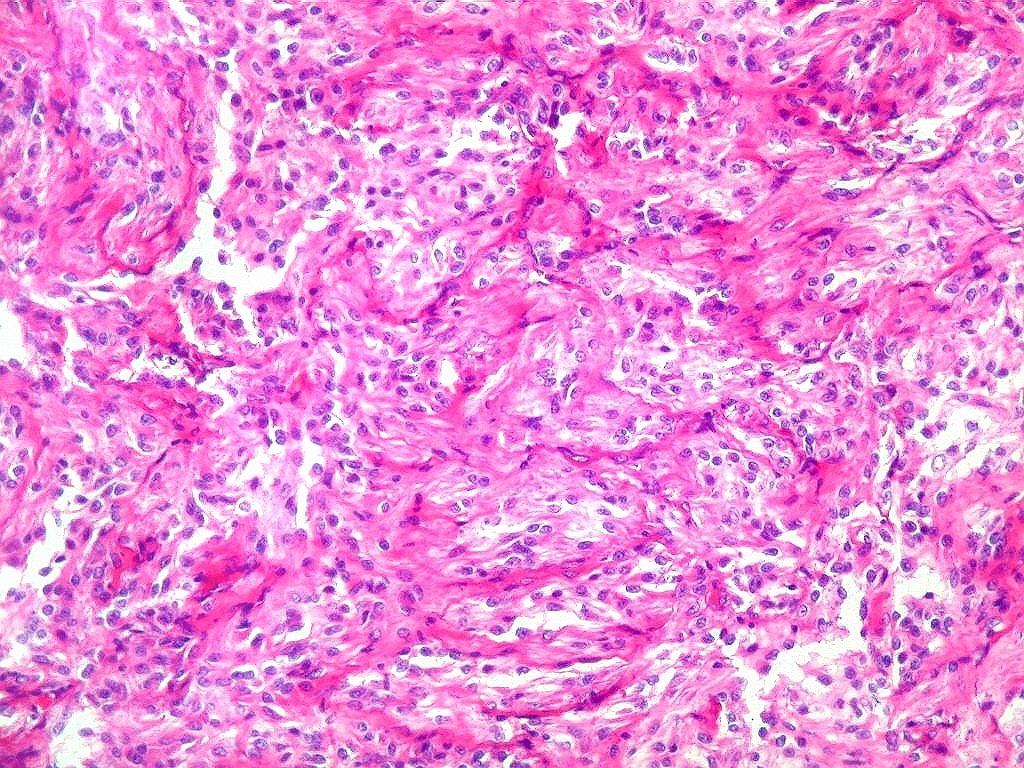
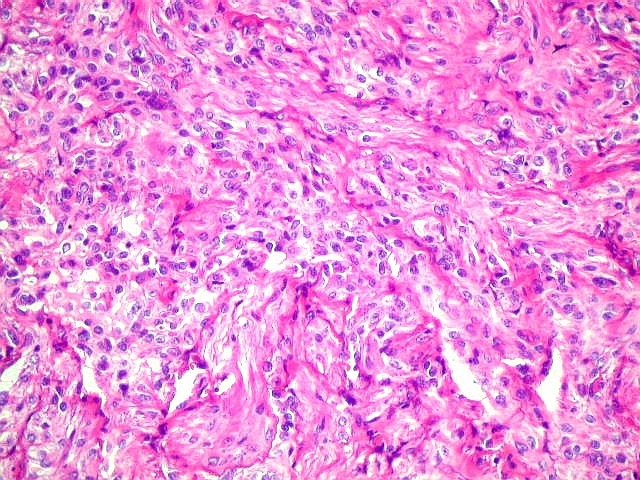
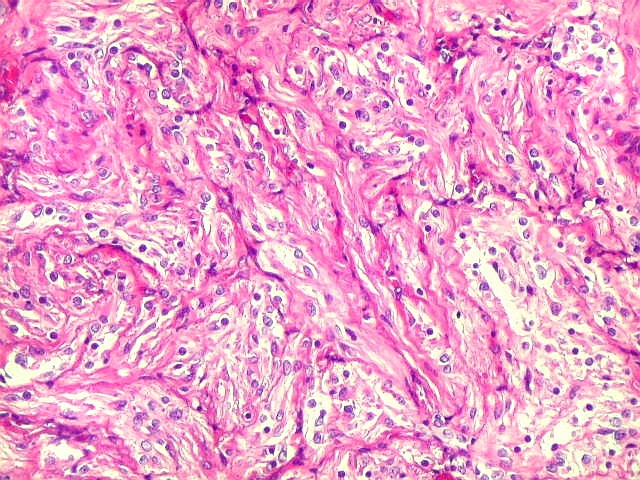
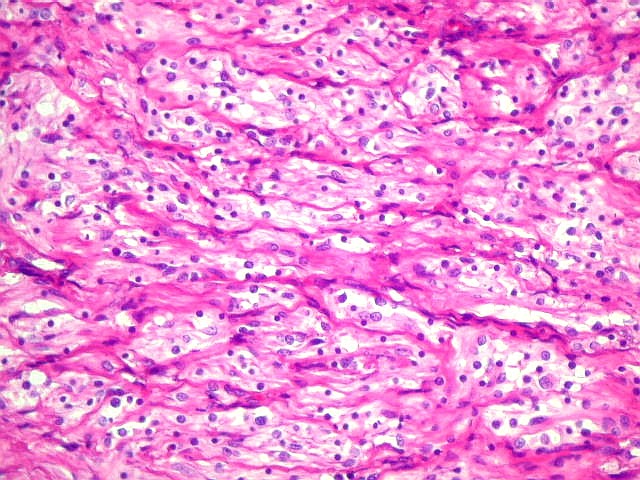
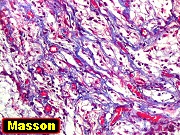
Micro.
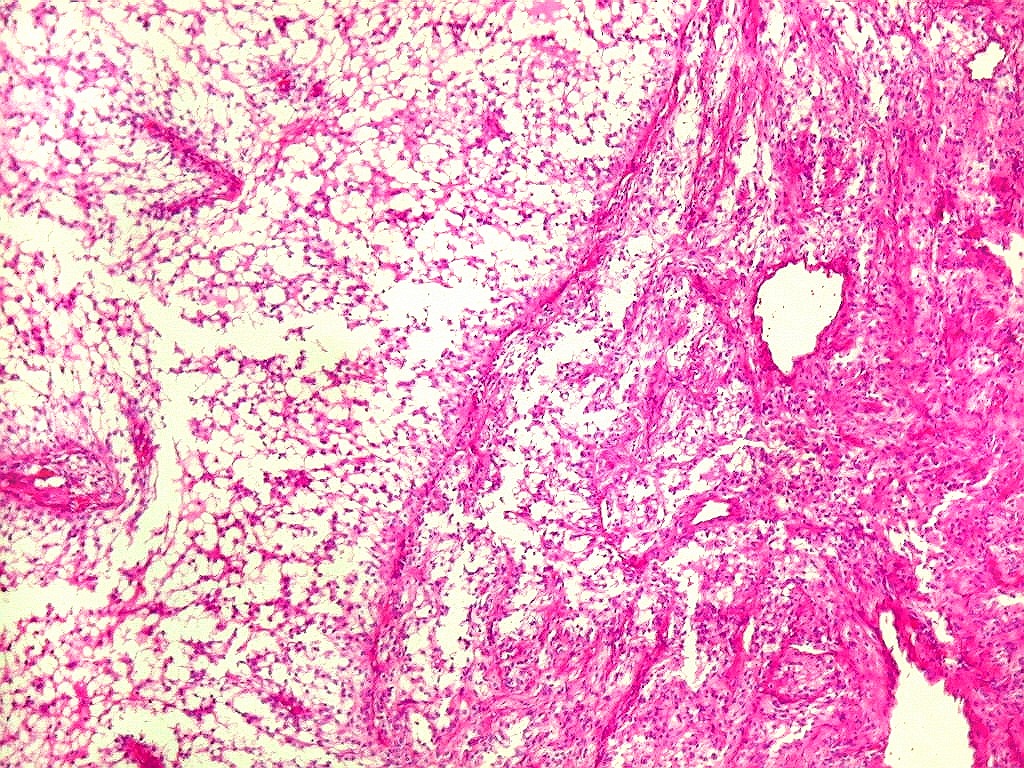
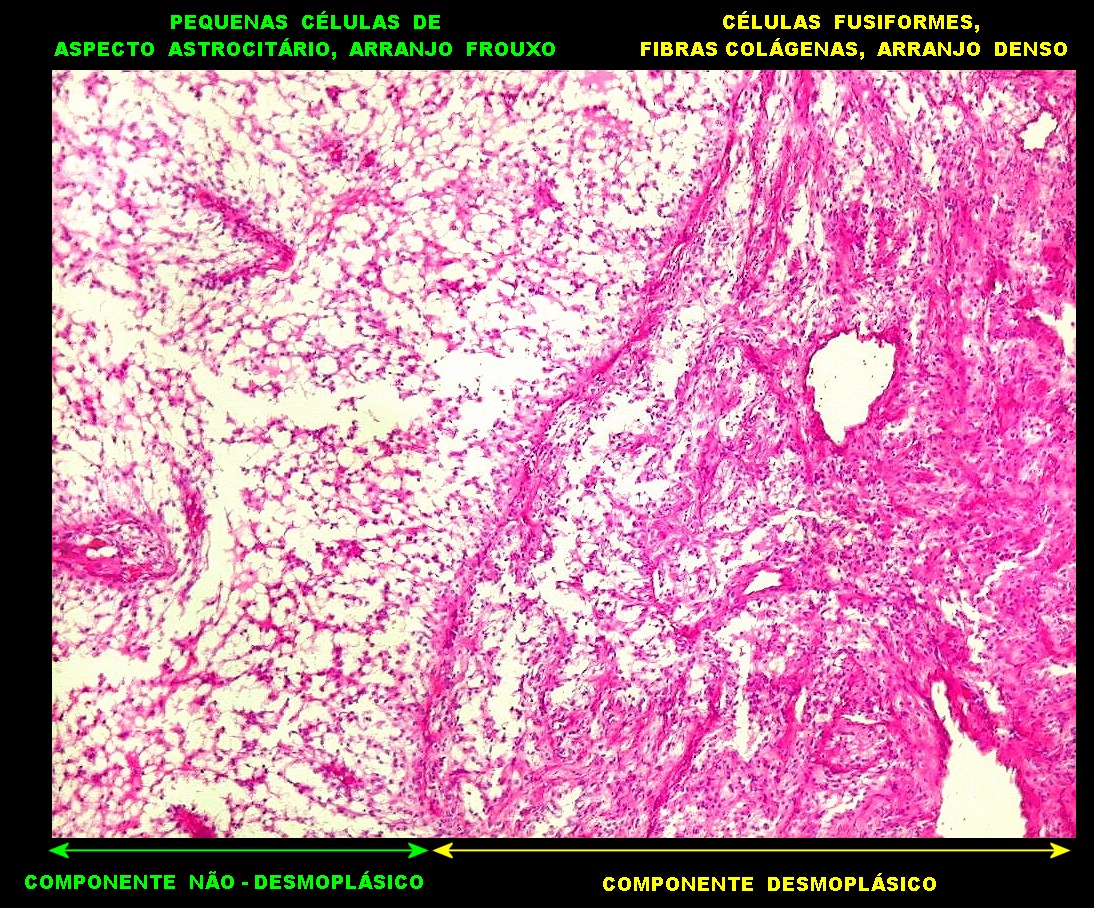
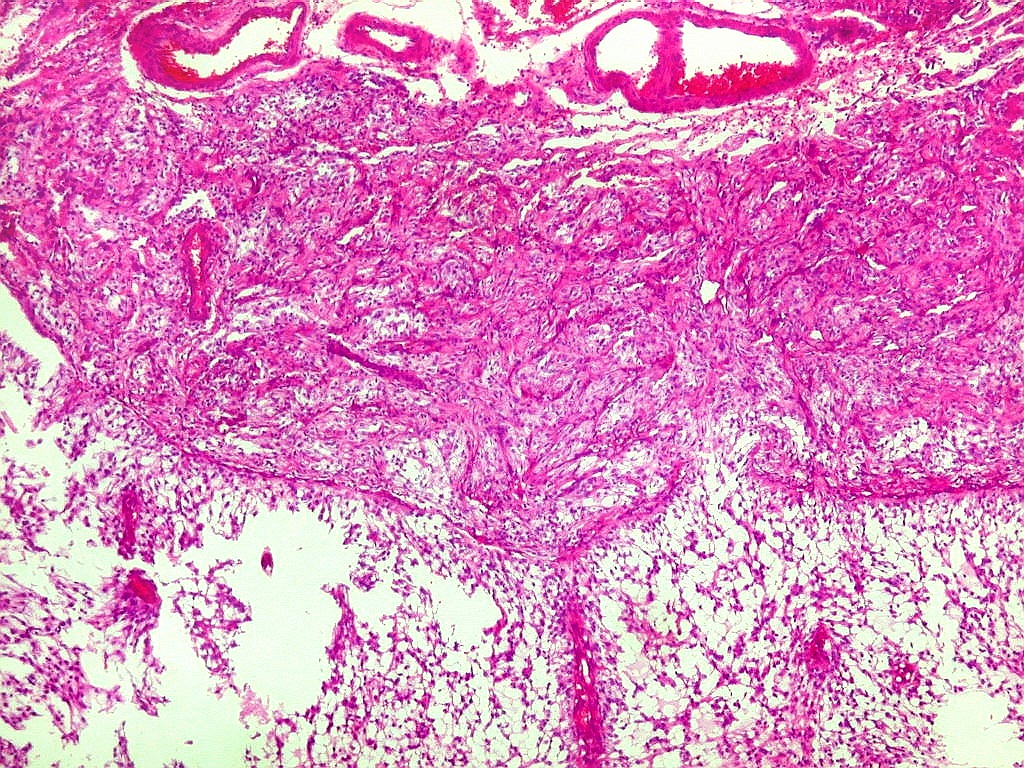
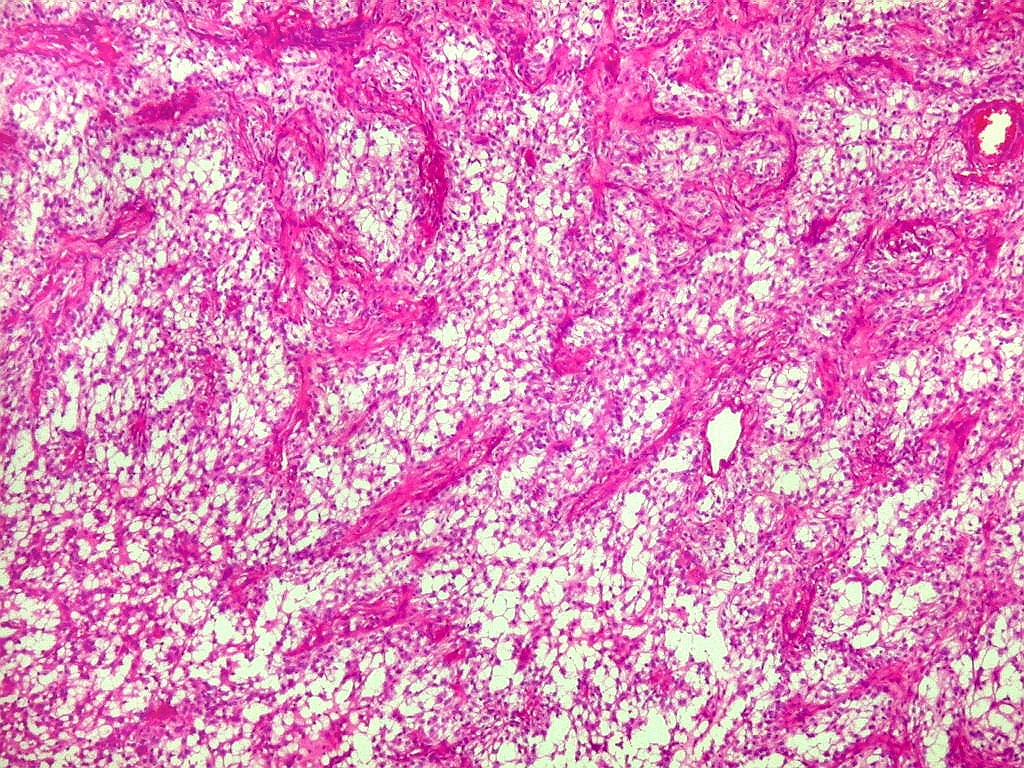
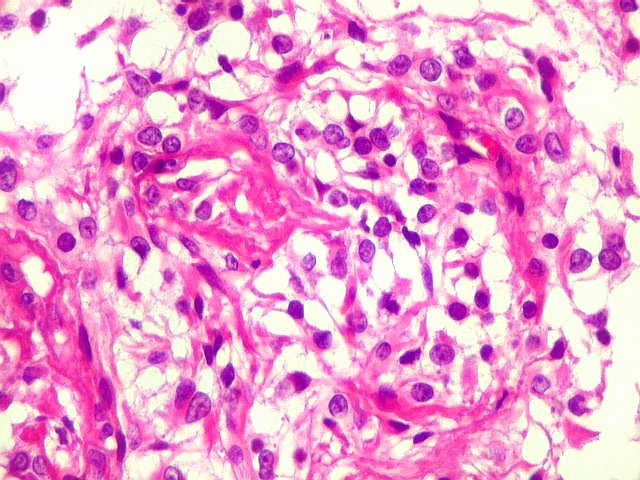
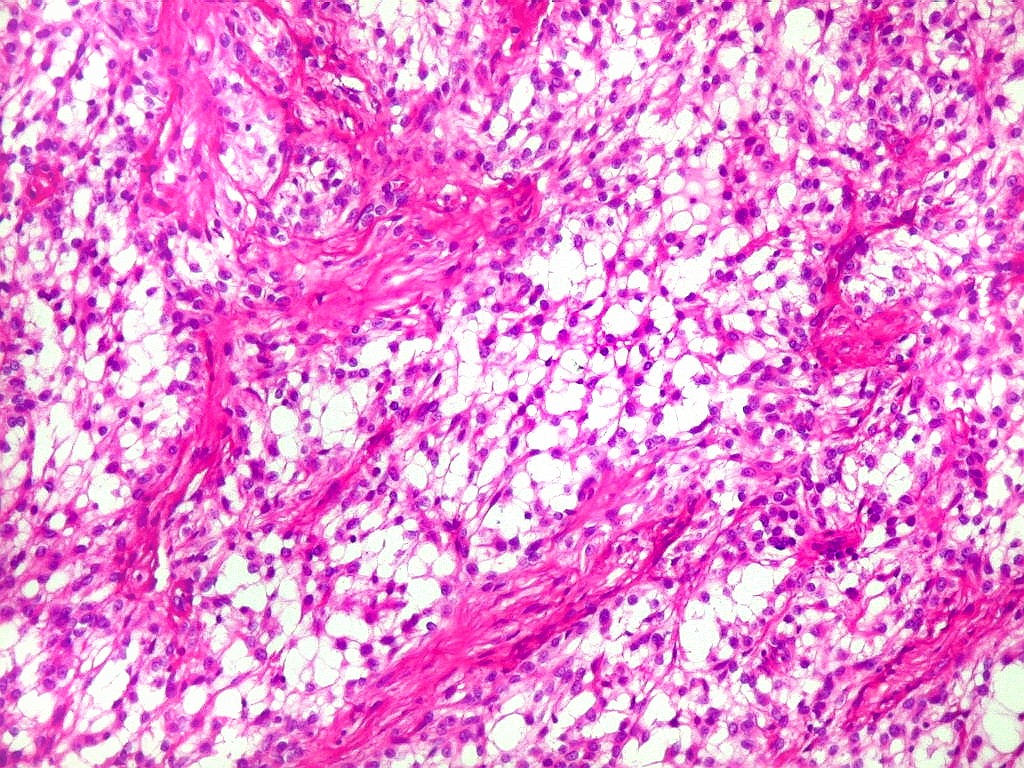
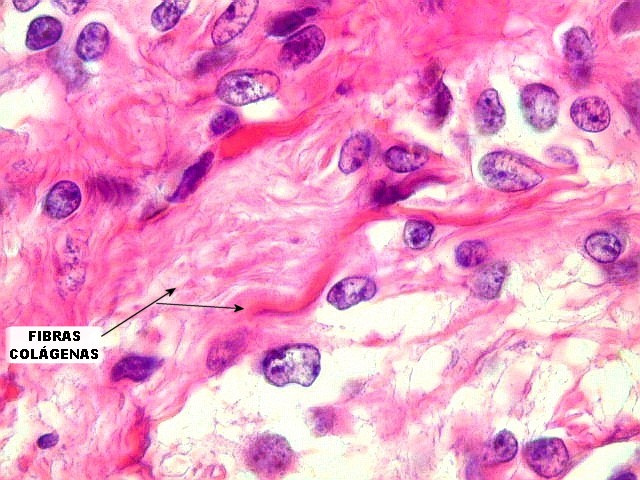
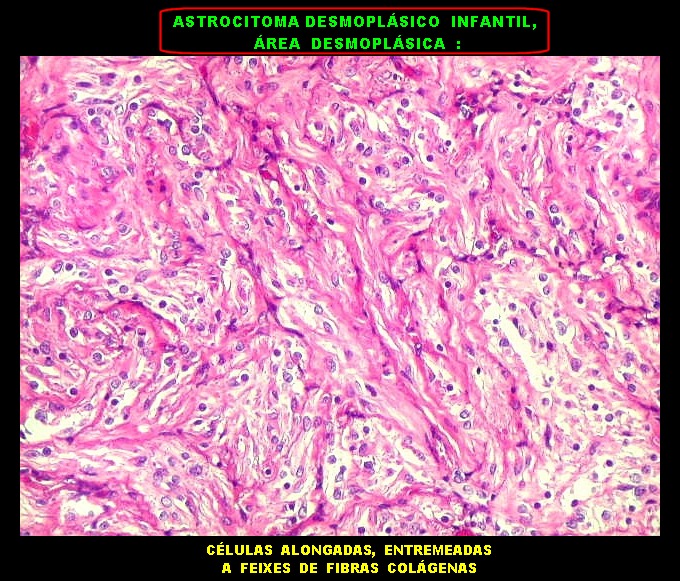
É característico um padrão bifásico, constituído
por uma porção desmoplásica leptomeníngea,
e um componente neuroepitelial pouco diferenciado. O componente leptomeníngeo
é formado por uma mistura de células fusiformes semelhantes
a fibroblastos e células neuroepiteliais pleomórficas arranjadas
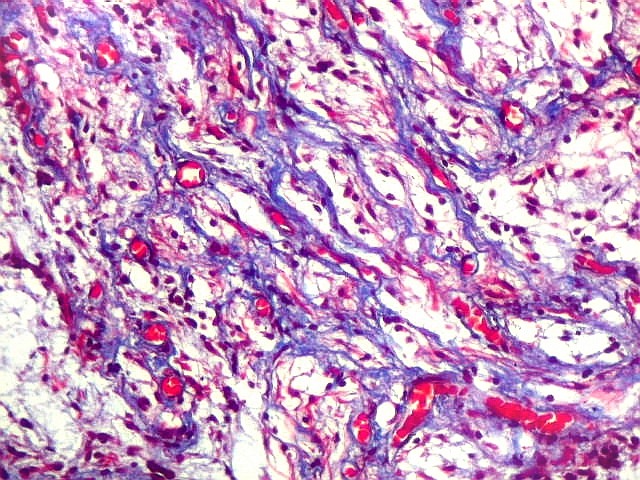
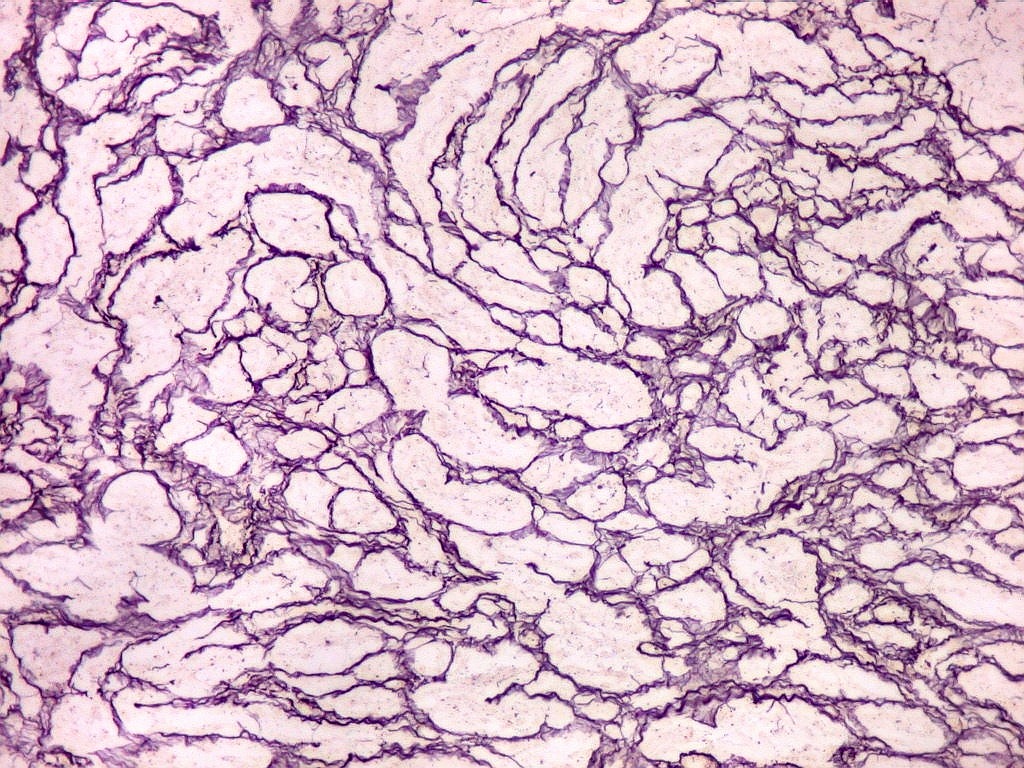
em feixes ou com padrão estoriforme. Impregnação
pela prata para reticulina mostra uma densa rede de fibras reticulínicas
envolvendo as células individualmente, como em uma neoplasia mesenquimal.
No astrocitoma desmoplásico infantil os astrócitos são
o único elemento neoplásico. No ganglioglioma desmoplásico
infantil há uma população variável de neurônios,
mas os astrócitos sempre predominam.
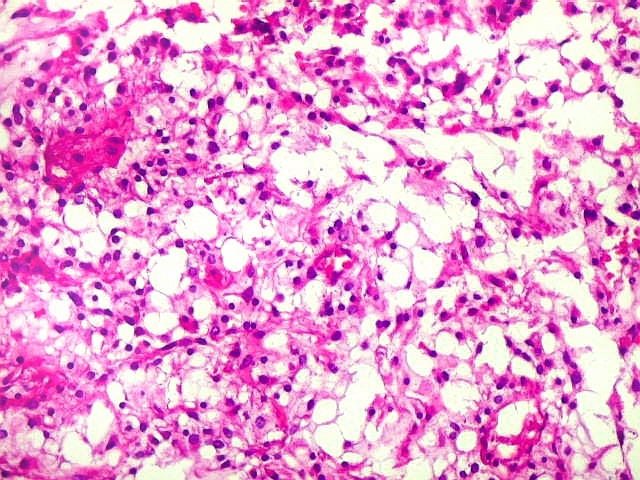
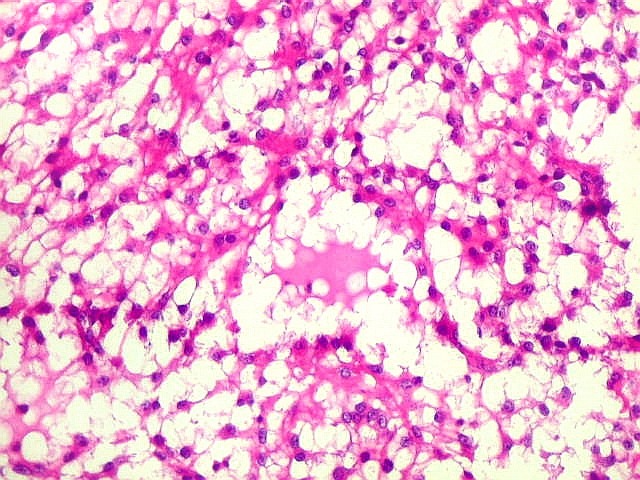
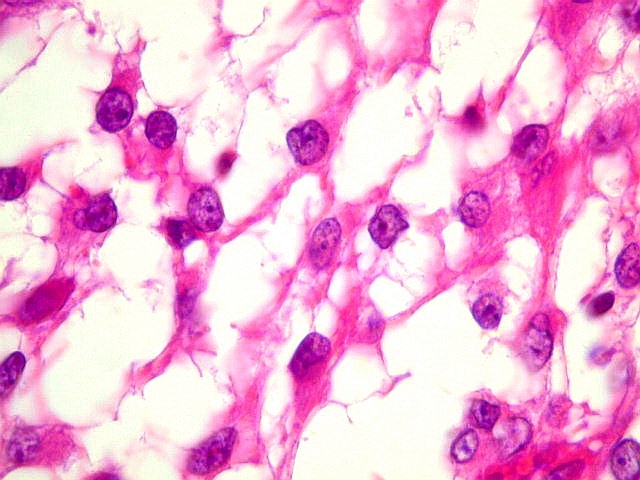
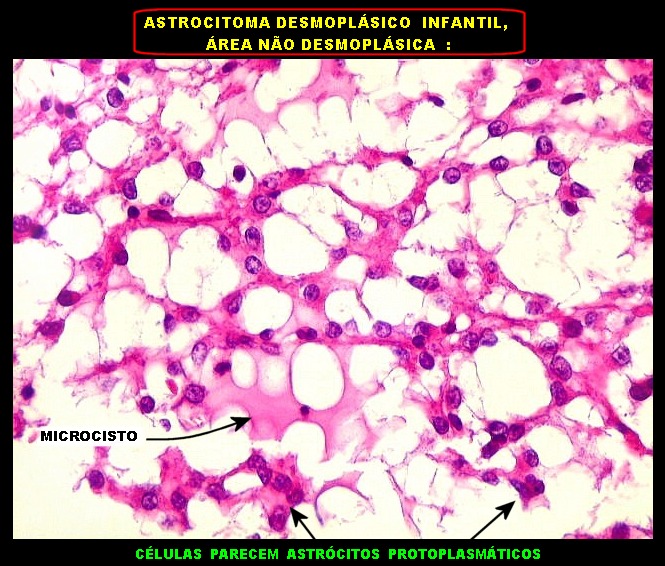
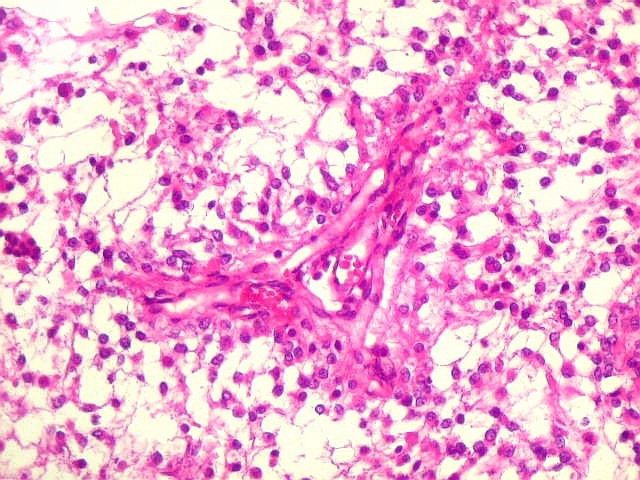
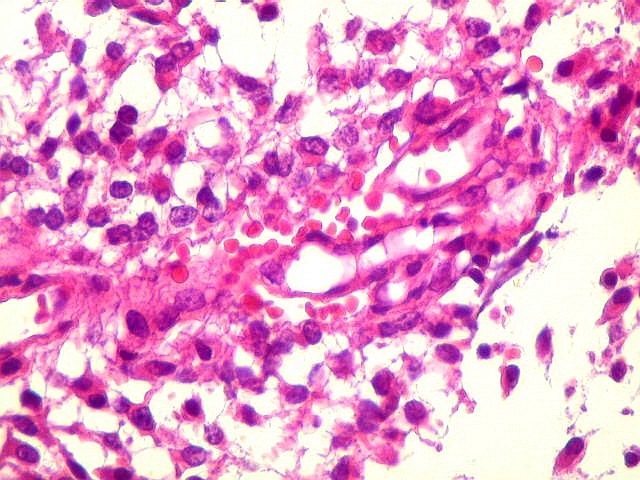
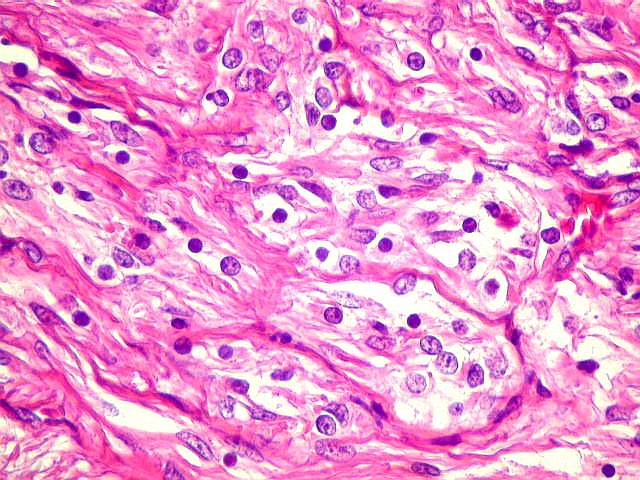
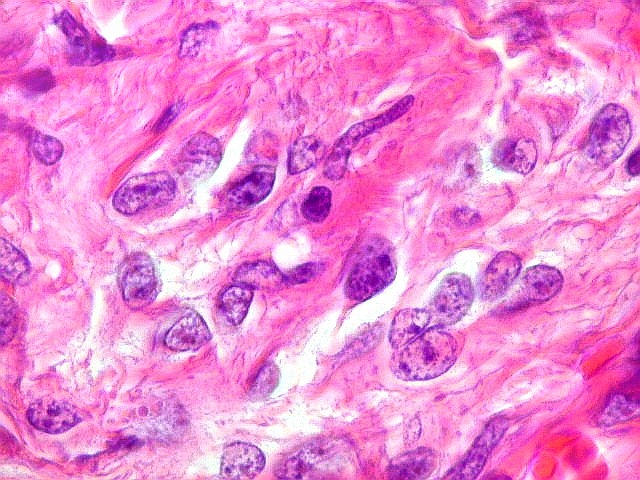
Além
deste componente desmoplásico, há uma população
de células neuroepiteliais pouco diferenciadas, com núcleos
pequenos redondos e de cromatina densa e citoplasma escasso. Tal
componente, sem desmoplasia, pode predominar em áreas e ter morfologia
astrocitária. Nestas áreas não há reticulina,
exceto associada a vasos. Há demarcação nítida
entre a superfície cortical e o tumor desmoplásico.
Geralmente não se observam mitoses, necrose ou proliferação
vascular.
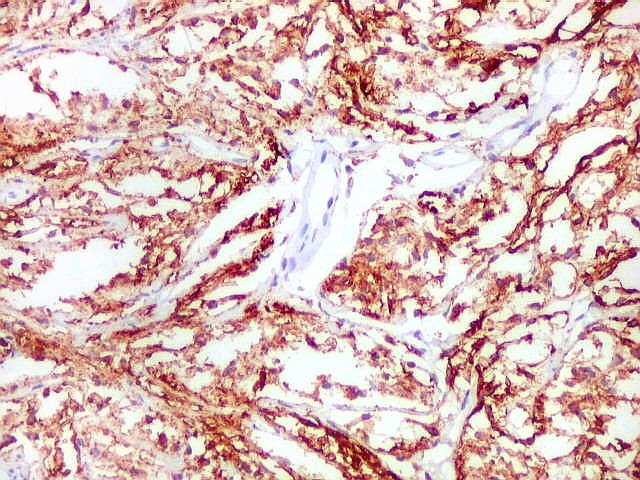
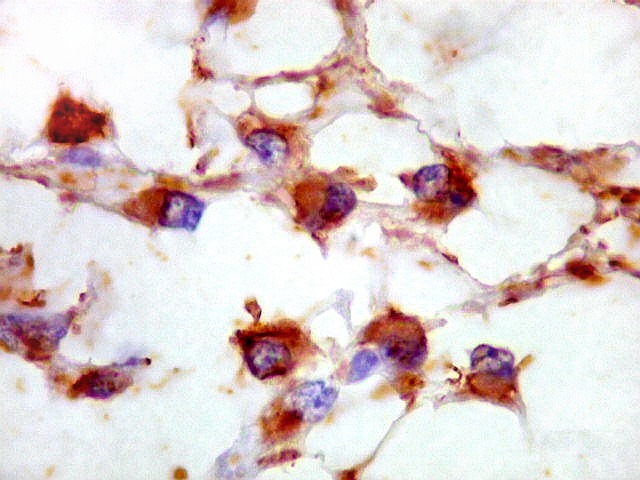
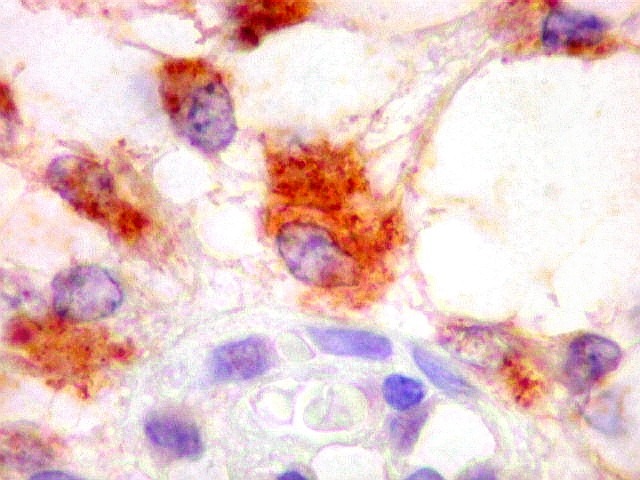
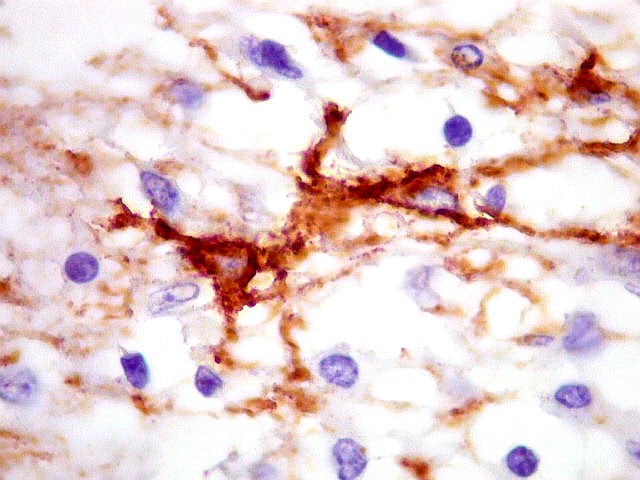
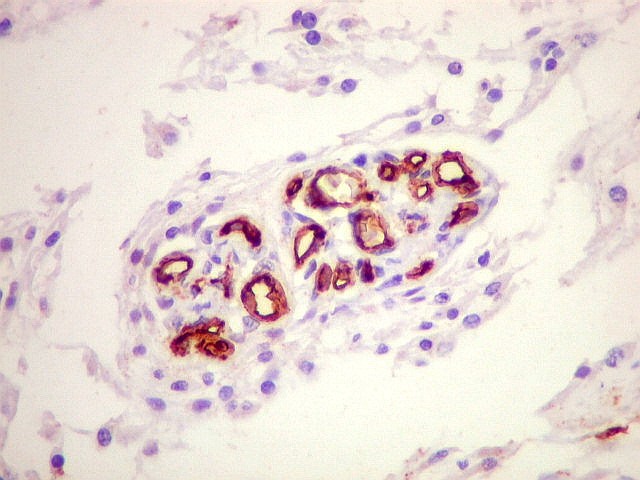
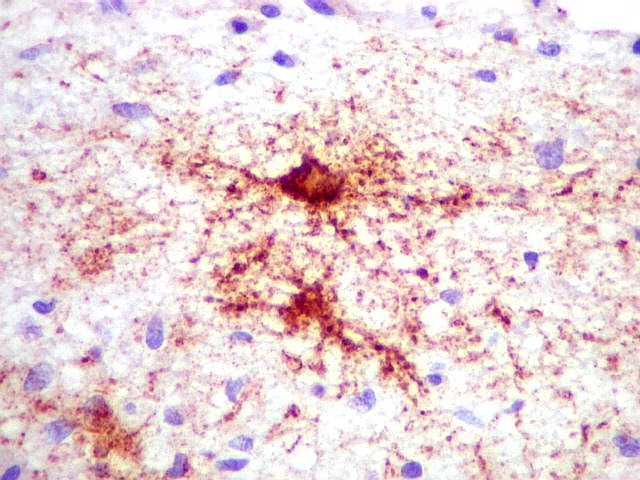
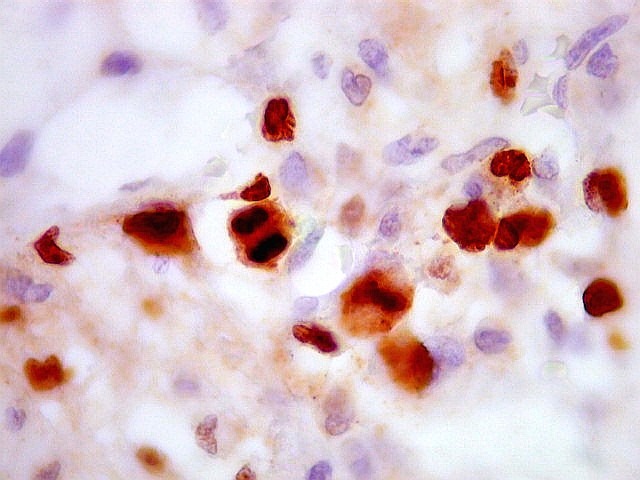
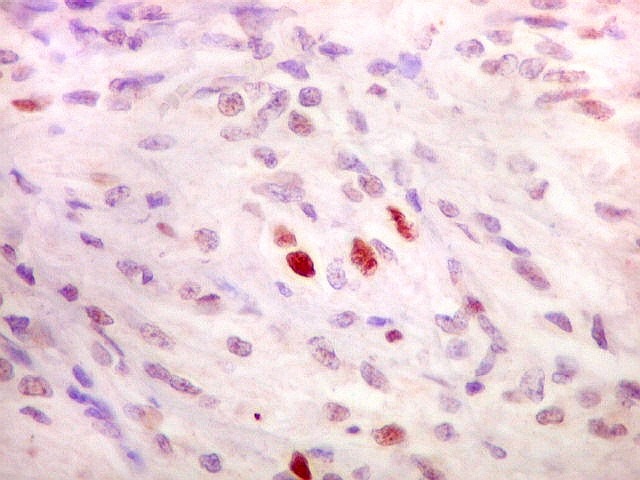
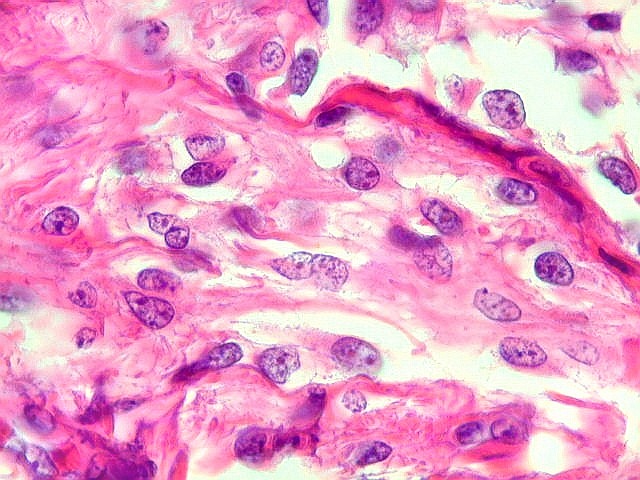
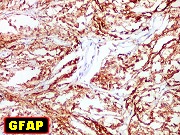
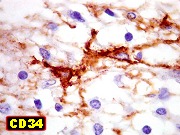
Imunohistoquímica.
As células neoplásicas, tanto no componente desmoplásico
como no não desmoplásico, marcam-se para GFAP e vimentina.
Há positividade para actina de músculo liso (1A4) em células
isoladas. Neurônios neoplásicos, quando presentes, expressam
marcadores neuronais como sinaptofisina e neurofilamento. Contudo, marcadores
neuronais podem também ser expressos pela população
de células pequenas pouco diferenciadas. Ki67 varia entre 0,5 e
5%, mas na maioria dos relatos fica abaixo de 2%. Há, porém,
casos raros com anaplasia, mitoses facilmente identificáveis e Ki67
de até 45%.
Histogênese.
A co-expressão de proteínas neuronais e gliais sugere que
as células dos DIA/DIGs sejam primitivas ou progenitoras. Assim,
estes tumores seriam embrionários e programados para diferenciação
progressiva. É possível que a origem dos mesmos seja
em astrócitos superficiais subpiais, especializados em formar membrana
basal. A escassa evidência genética disponível sugere
que os DIA/DIGs não sejam relacionados aos astrocitomas difusos
comuns.
Prognóstico.
Ressecção macroscopicamente completa resulta em sobrevida
longa. Em um estudo de 14 pacientes, a sobrevida média foi 8,7 anos
(1 a 14 anos). Quando há remoção subtotal ou só
biópsia, a evolução é lenta, descrevendo-se
até estabilidade ou regressão. O encontro de mitoses ou necrose
não parece afetar o bom prognóstico.
Fontes:
Brat DJ,
VandenBerg SR, Figarella-Branger D, Taratuto AL. Desmoplastic infantile
astrocytoma and ganglioglioma. In WHO Classification
of Tumours of the Central Nervous System. 4th Ed. Louis DN, Ohgaki
H, Wiestler OD, Cavenee WK, editors. International Agency for Research
on Cancer, Lyon, 2007. pp 96-8.
Brat DJ.
Neuronal and Glioneuronal Neoplasms. In Practical Surgical
Neuropathology. Perry A, Brat DJ, editors. Churchill Livingstone
Elsevier, 2010. pp 130-3. |