|
|
|
|
|
|
|
|
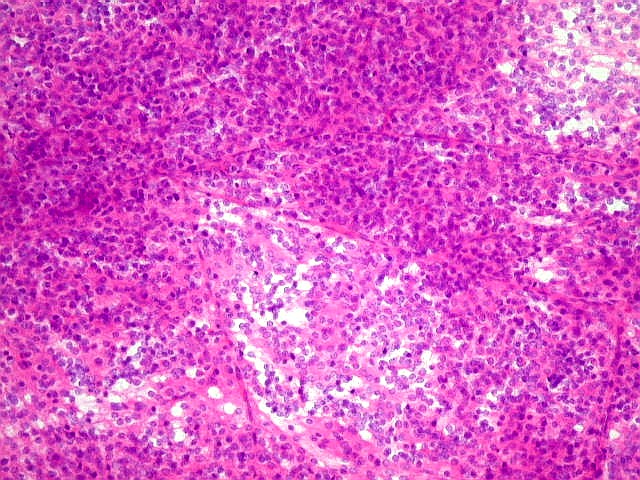
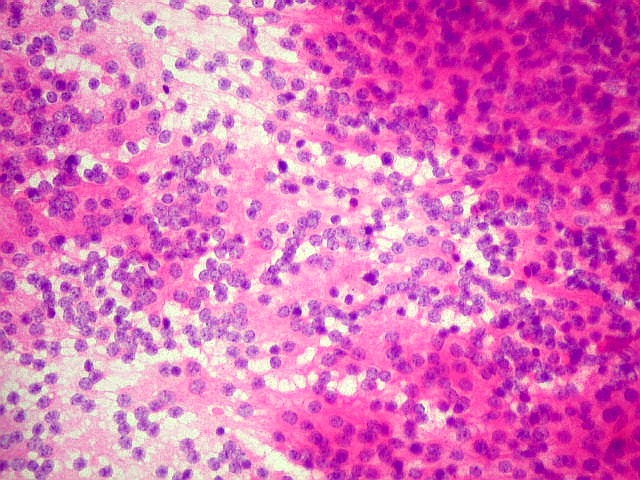
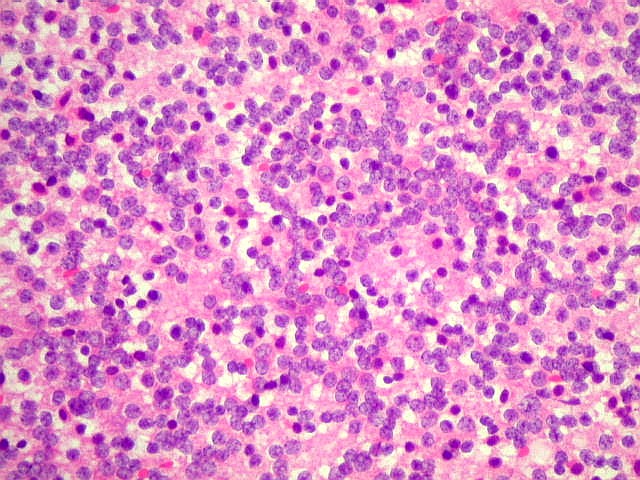
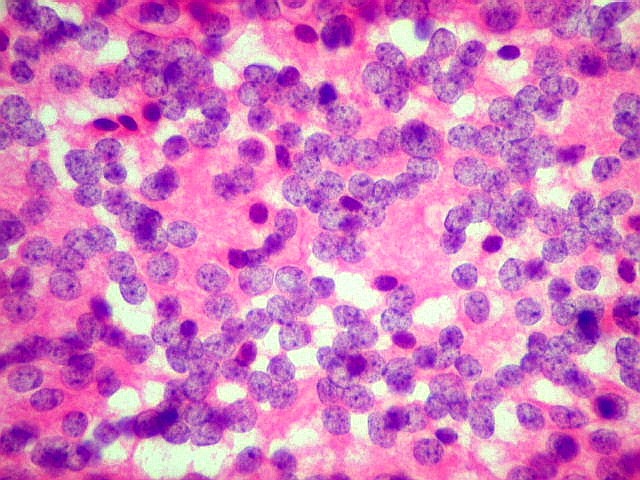
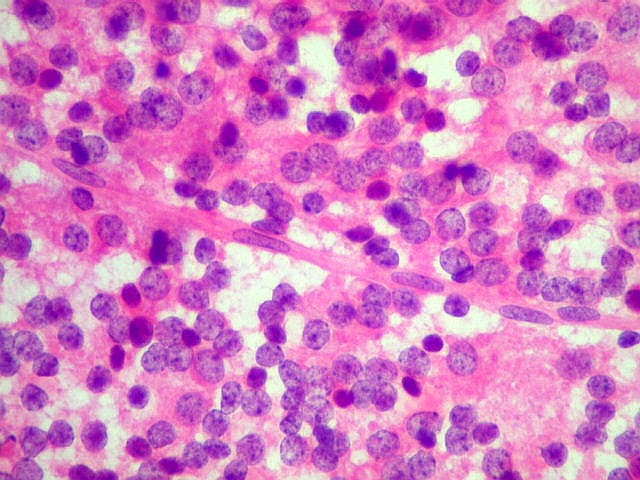
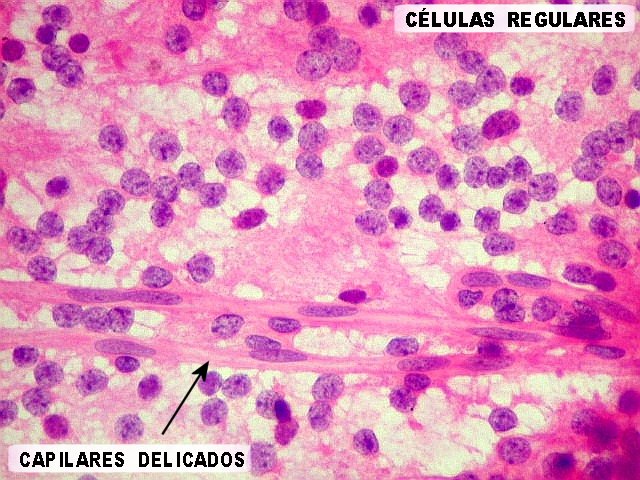
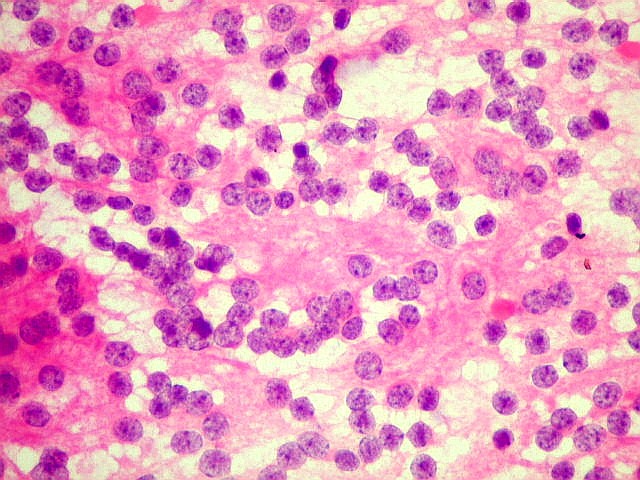
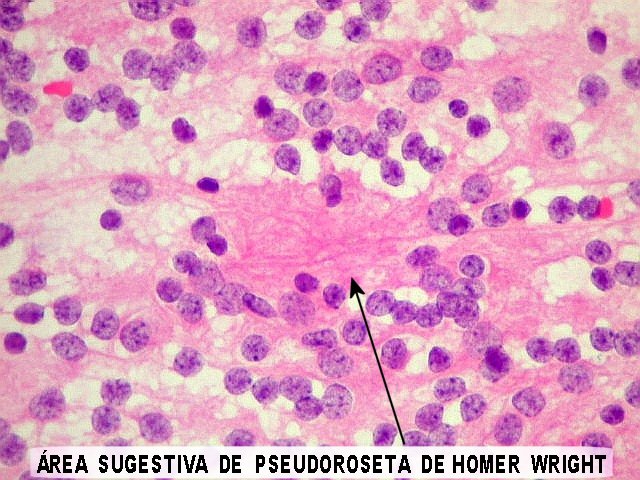
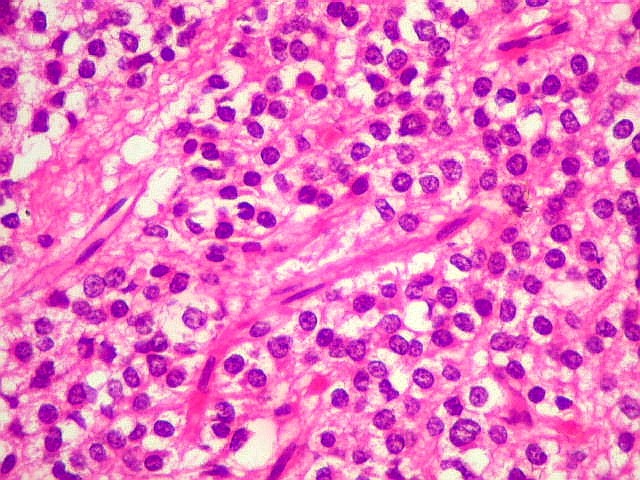
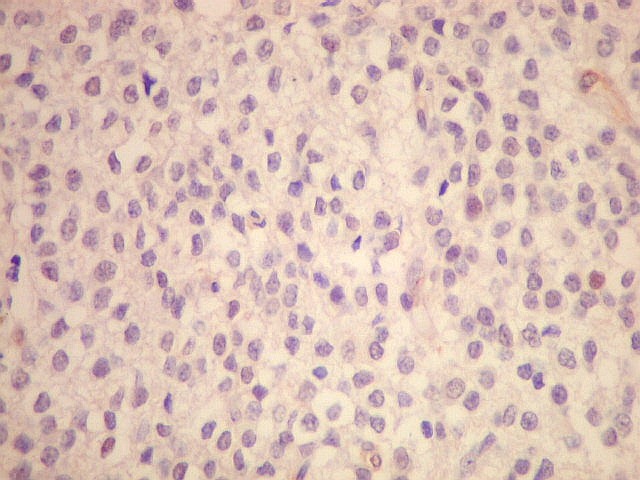
| Esfregaço. Pequena amostra do tumor a fresco, esfregada entre duas lâminas e corada por HE, mostra neoplasia altamente celular, em arranjo sólido, composta por células com núcleos arredondados e isomorfos, cromatina frouxa, e citoplasma róseo de limites indistintos. Os capilares são delicados, com núcleos de células endoteliais espaçados, sem proliferação. Ocasionalmente, há focos de tecido anucleado, contendo o que parecem ser prolongamentos das células neoplásicas circundados por núcleos, lembrando as chamadas pseudorosetas de Homer Wright, uma feição sugestiva de diferenciação neuronal. | |
 |
 |
 |
 |
 |
 |
 |
 |
|
|
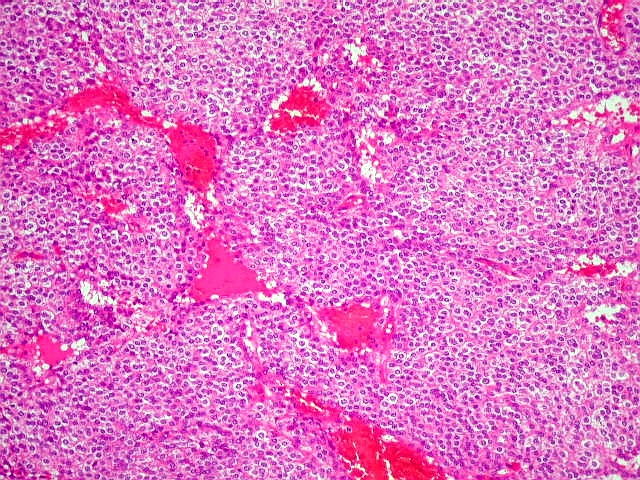
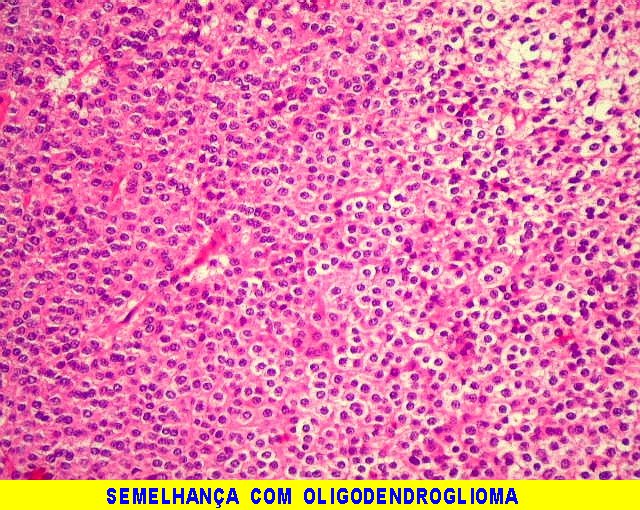
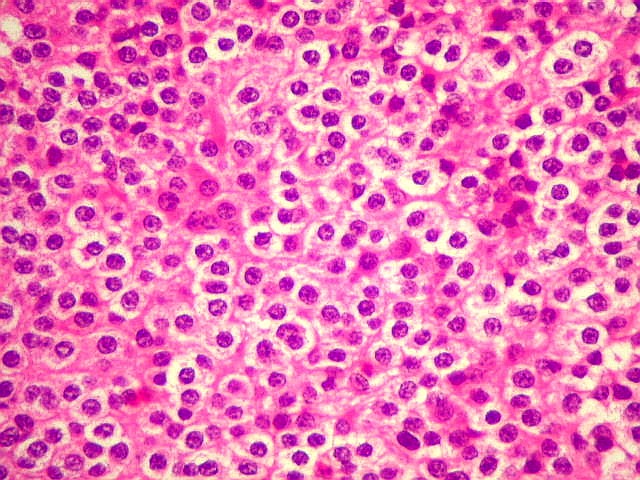
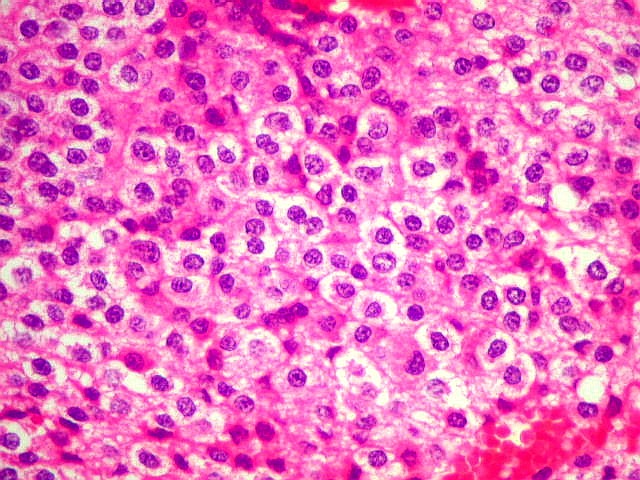
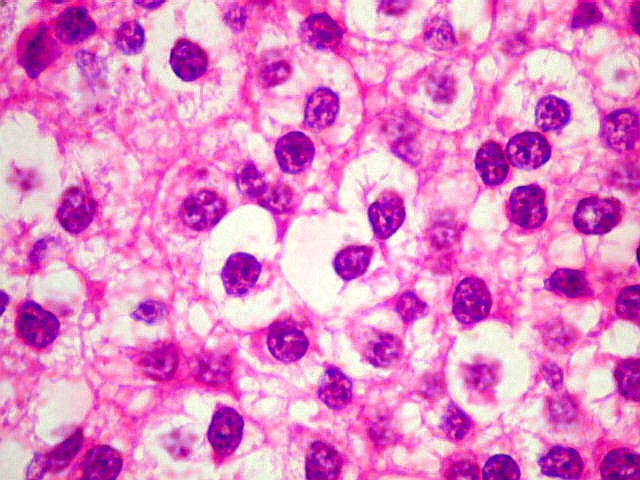
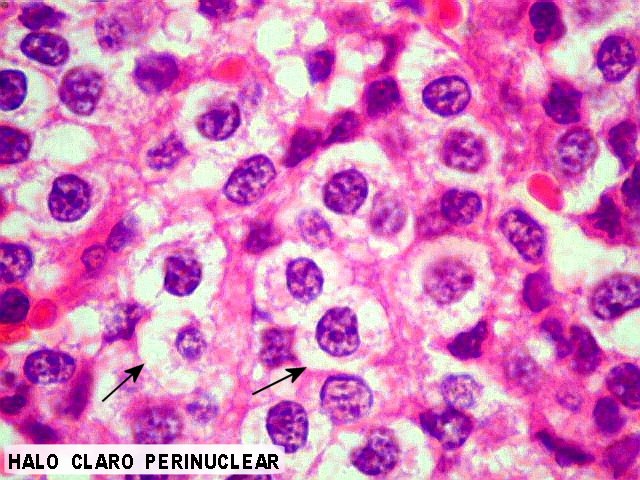
| Aspecto geral. É semelhante ao já descrito para o esfregaço. Chama a atenção a grande regularidade das células, com seus núcleos redondos e halo claro perinuclear (aspecto 'em ovo frito'), lembrando fortemente um oligodendroglioma. Não se observam proliferação vascular, áreas de necrose ou figuras de mitose. As imagens sugestivas de pseudorosetas de Homer Wright, notadas no esfregaço, não se repetem aqui. | |
 |
|
 |
 |
 |
 |
 |
 |
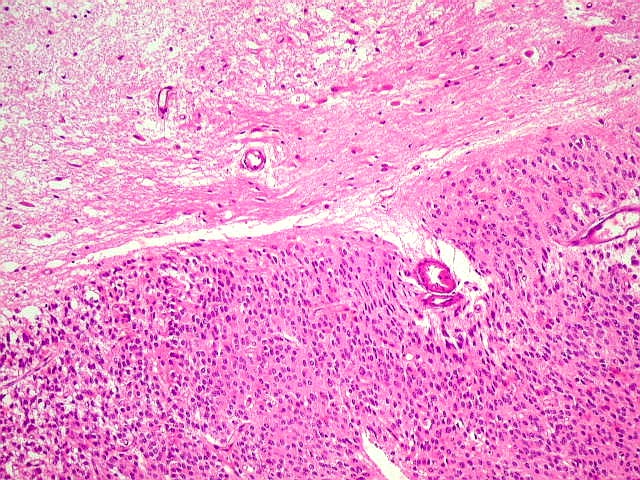
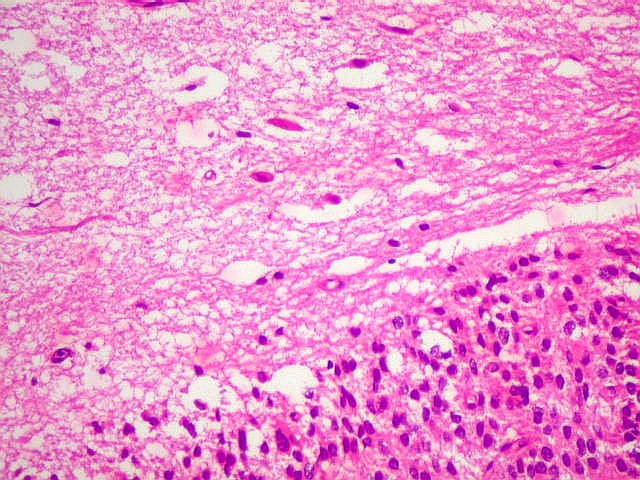
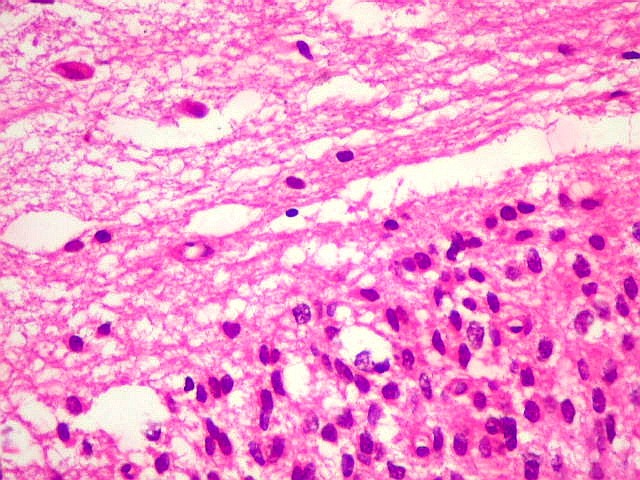
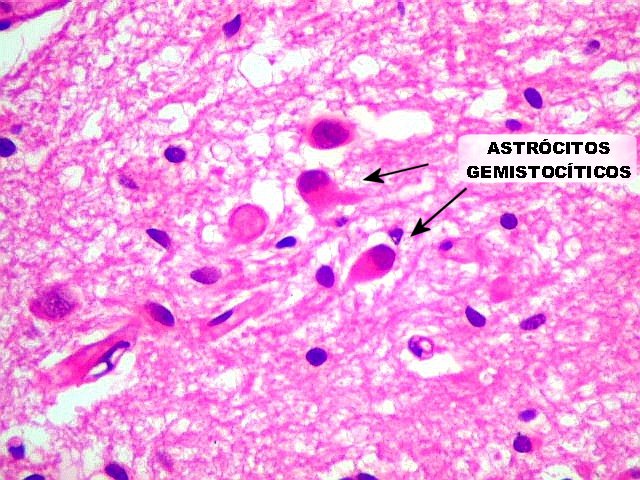
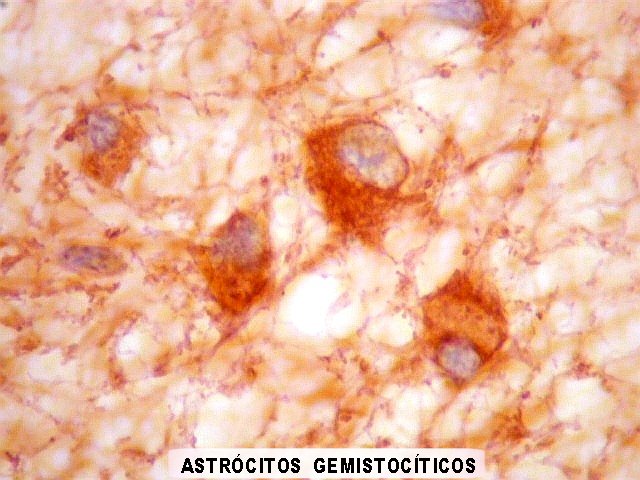
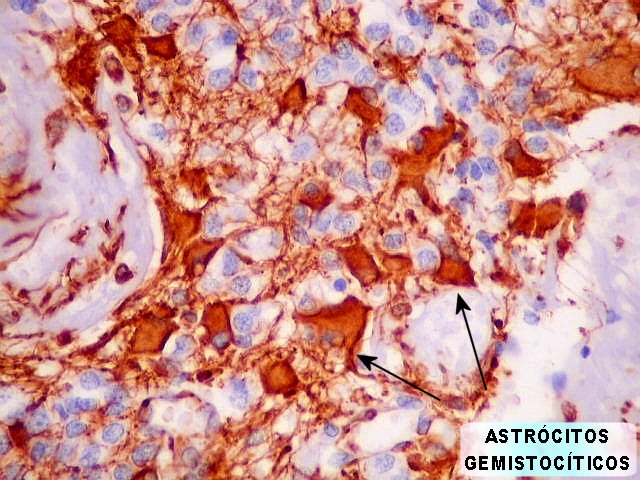
| Limite externo do tumor. A interface entre o tumor e o cérebro é nítida, embora não haja cápsula. No tecido vizinho há astrócitos hipertróficos reativos (gemistocíticos). | |
 |
 |
 |
 |
| IMUNOHISTOQUÍMICA |
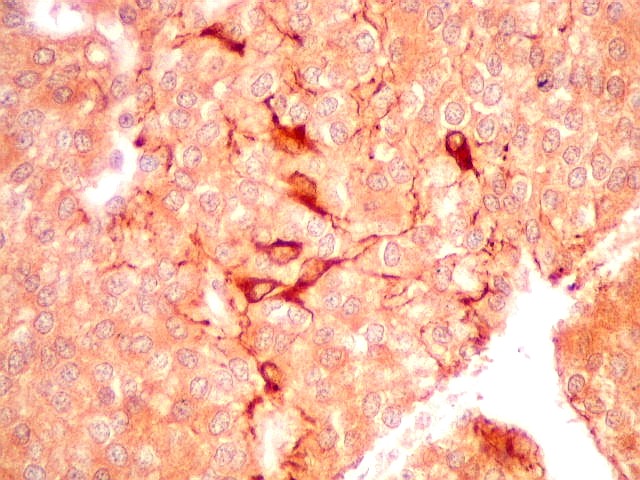
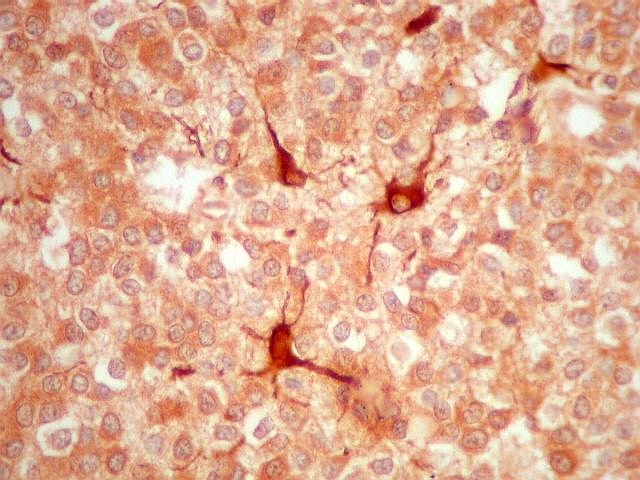
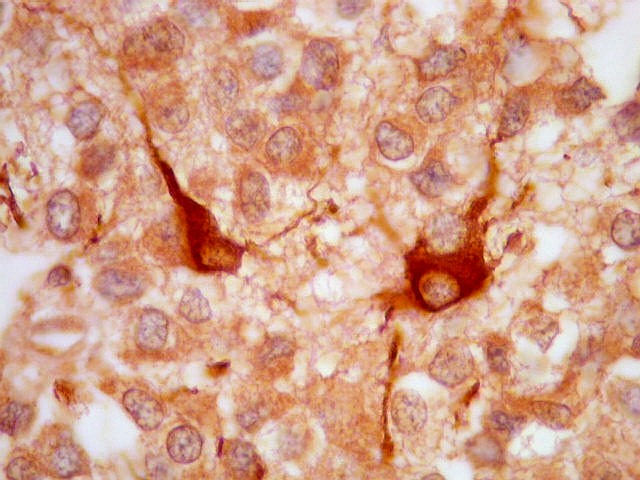
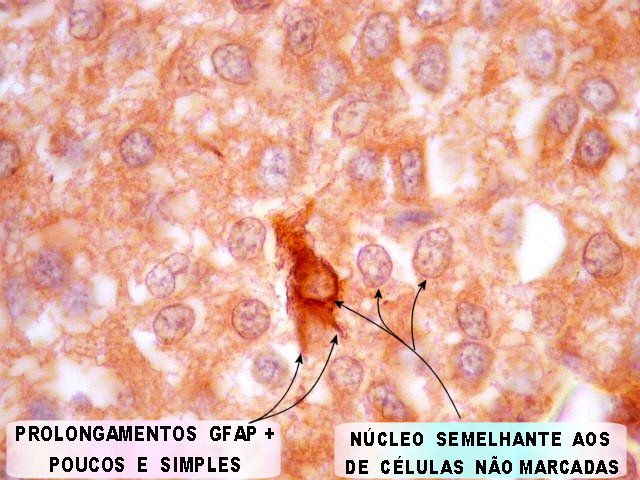
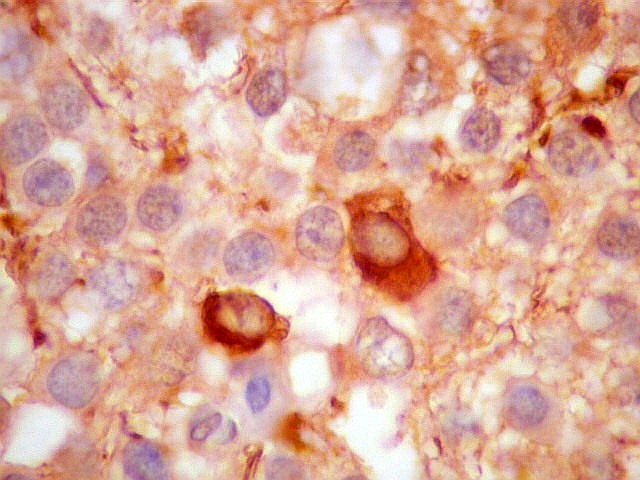
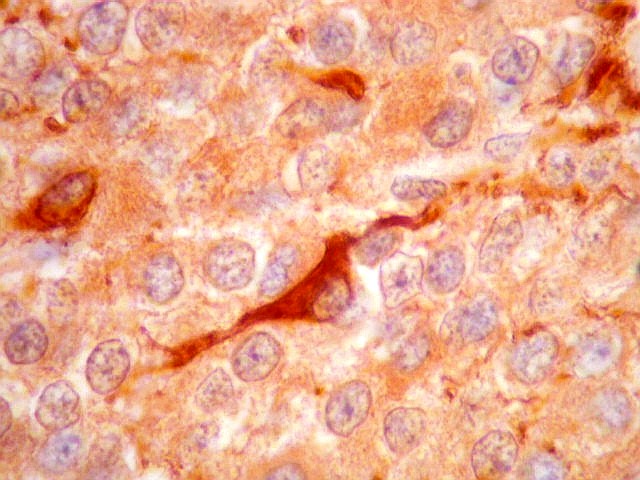
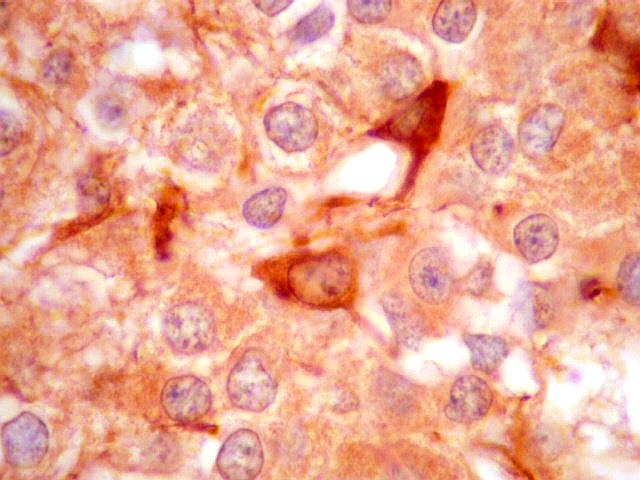
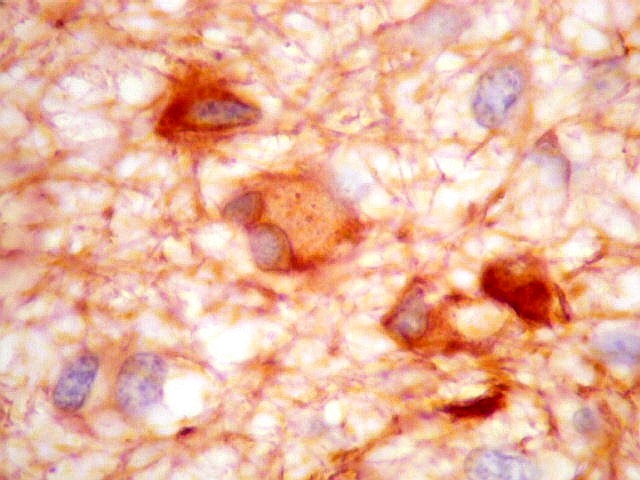
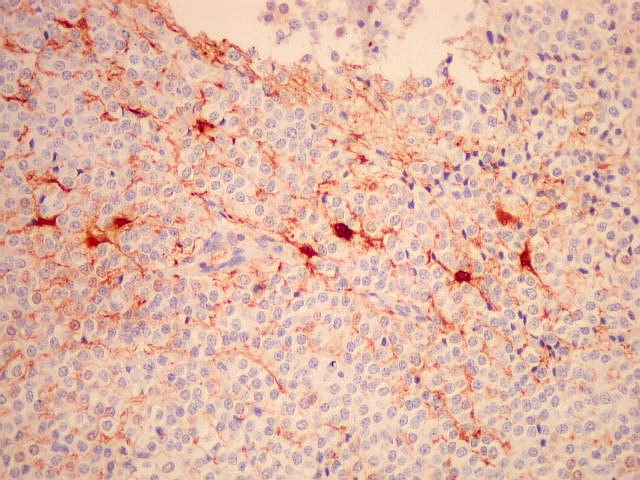
| GFAP. Há marcação de muitas células no interior do tumor, longe de áreas limítrofes com cérebro. A marcação é citoplasmática, mostrando delicados prolongamentos. Os núcleos, que não se coram, são de tamalho e características semelhantes aos das células não marcadas, sugerindo que as células positivas são também neoplásicas. O caráter simples dos prolongamentos, curtos e com poucas ramificações, apóia esta hipótese. | |
 |
 |
 |
 |
 |
 |
 |
 |
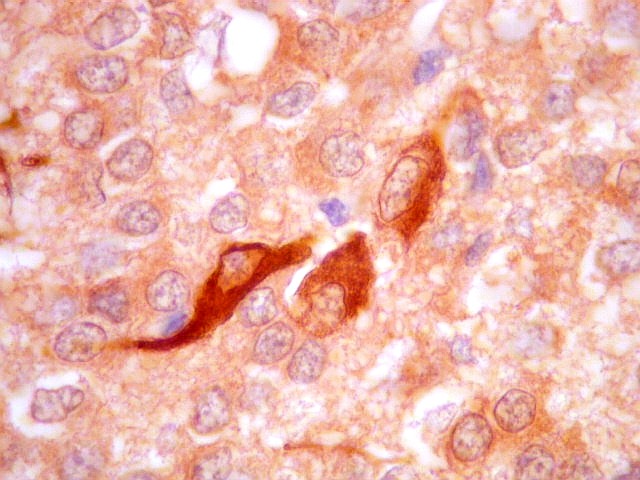
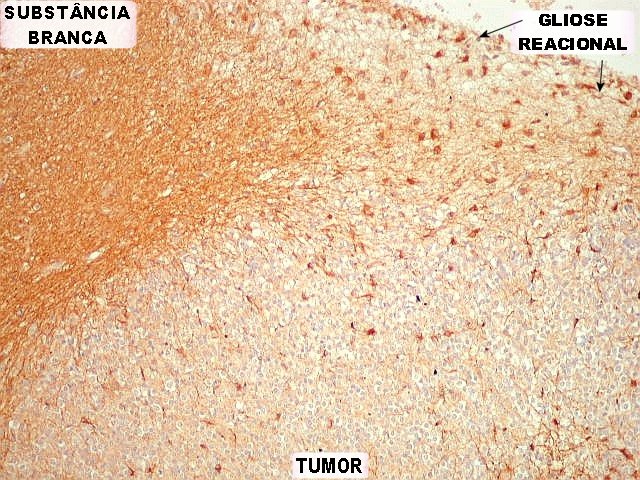
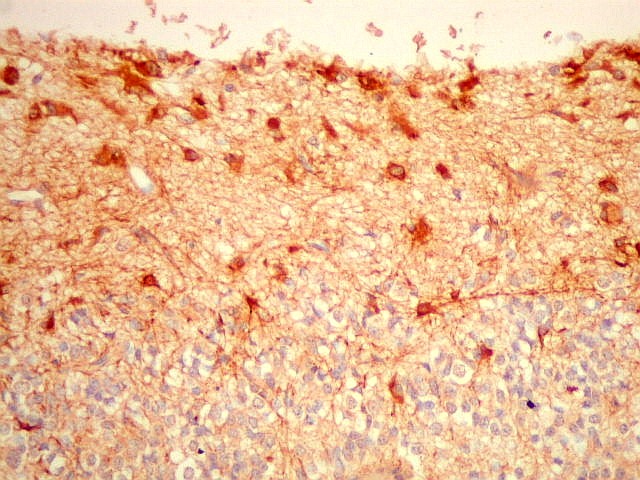
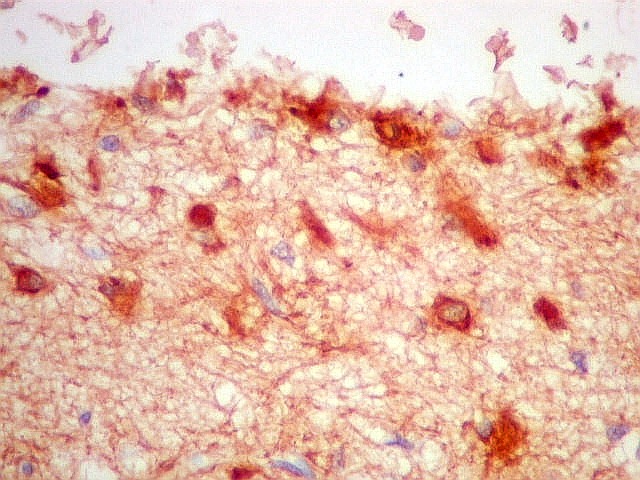
| GFAP - Periferia do tumor. Aqui observam-se vários astrócitos reativos (gliose), maiores que os demonstrados no interior do tumor. O núcleo é maior, excêntrico, o citoplasma mais abundante, com prolongamentos mais elaborados e ramificados. | |
 |
 |
 |
 |
 |
 |
 |
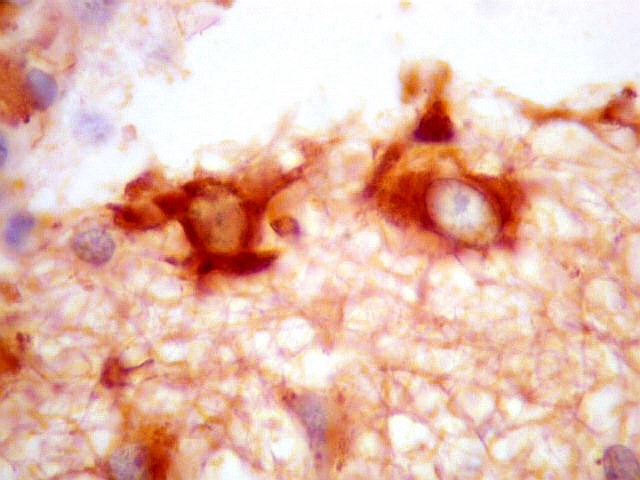 |
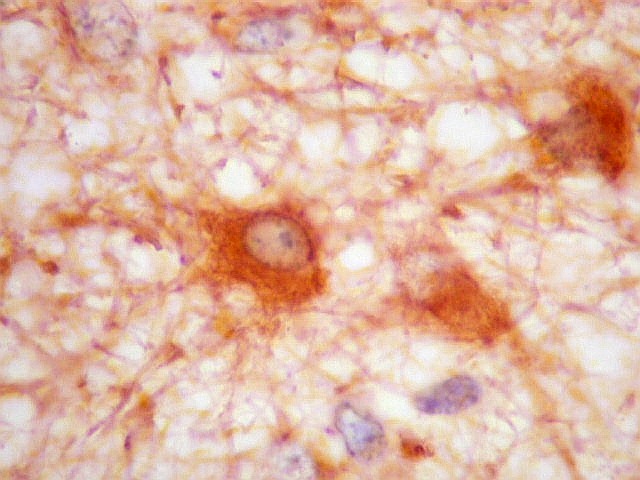
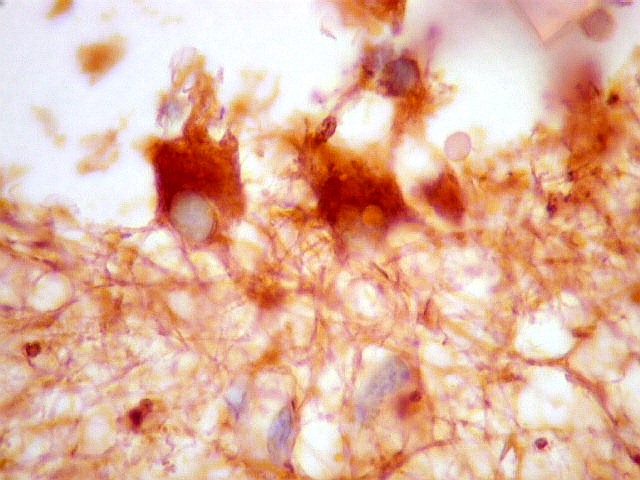
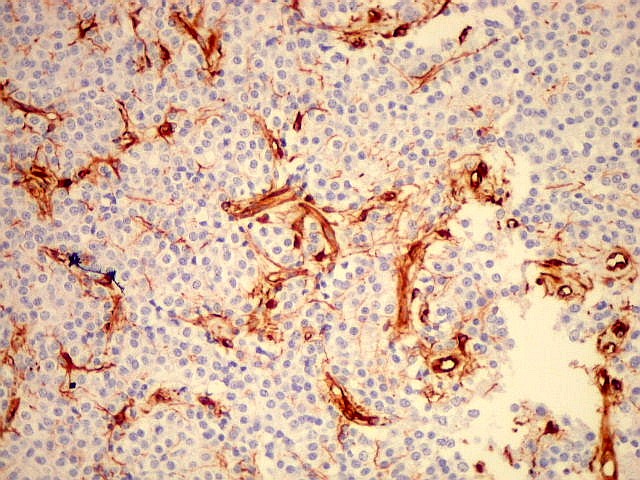
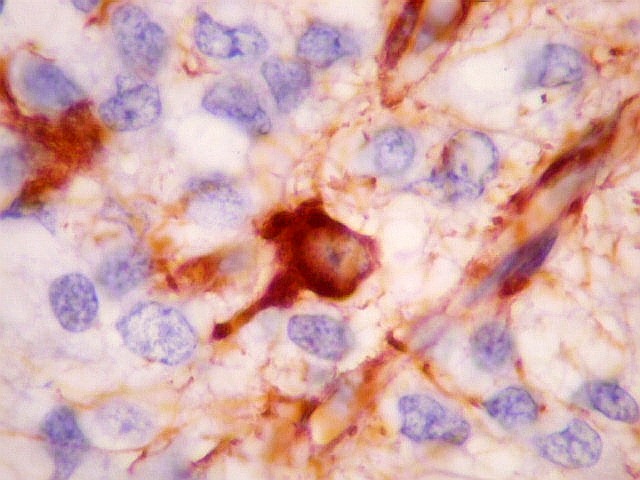
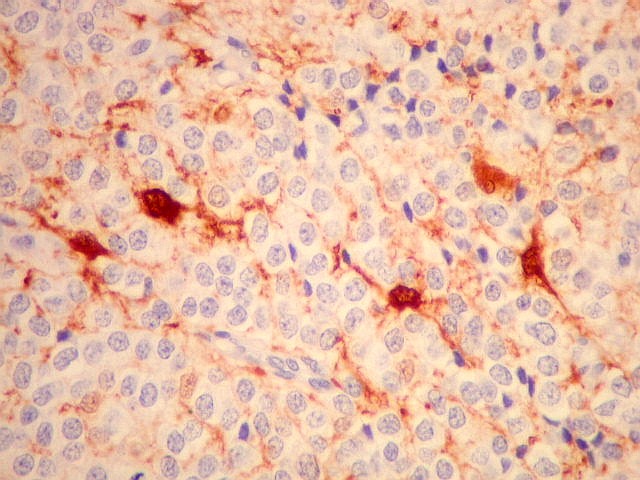
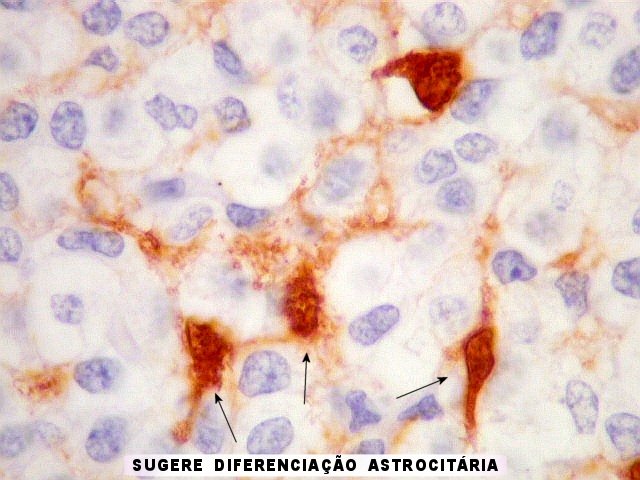
| VIM. Vimentina marca vasos (inclusive capilares), macrófagos (não aparentes em HE), astrócitos gemistocíticos reacionais da periferia do tumor, e células neoplásicas com presumível diferenciação astrocitária. A grande maioria das células neoplásicas não são marcadas. Em aumento fraco destaca-se a rede capilar. Com aumento mais forte, notam-se células neoplásicas reativas, com prolongamentos escassos e pouco ramificados, como já notado com GFAP. São interpretadas como células neoplásicas com diferenciação astrocitária. | |
 |
|
 |
 |
 |
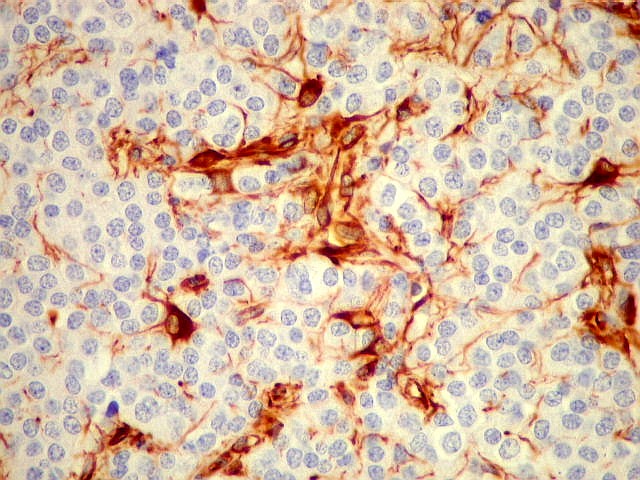 |
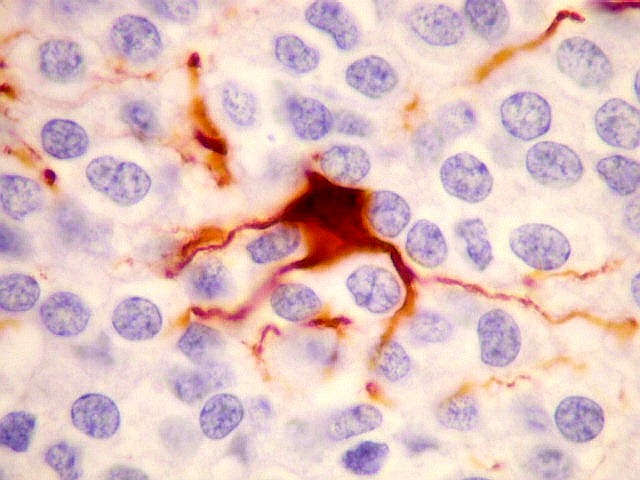
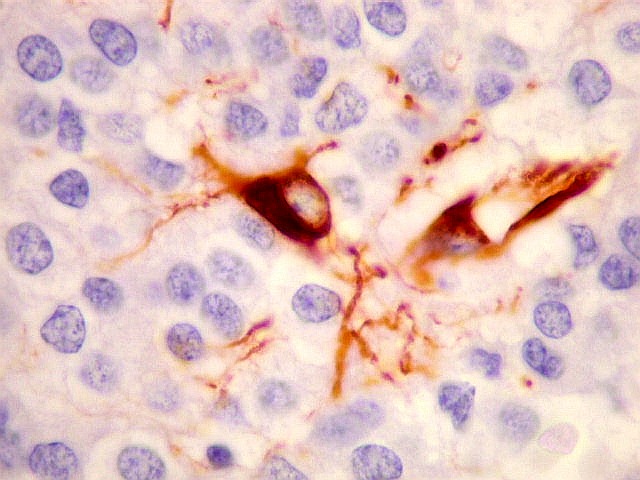
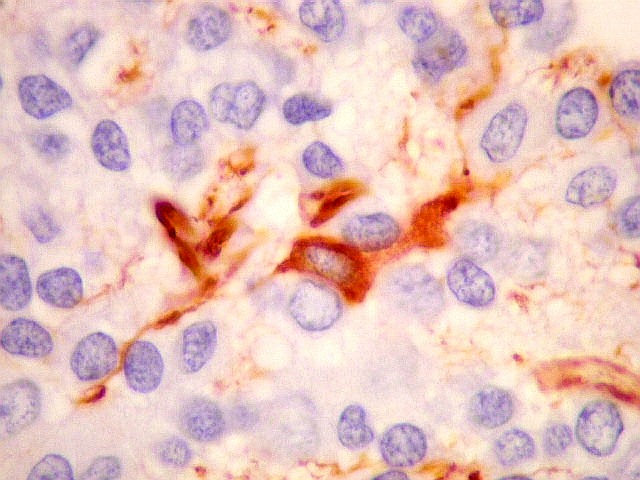
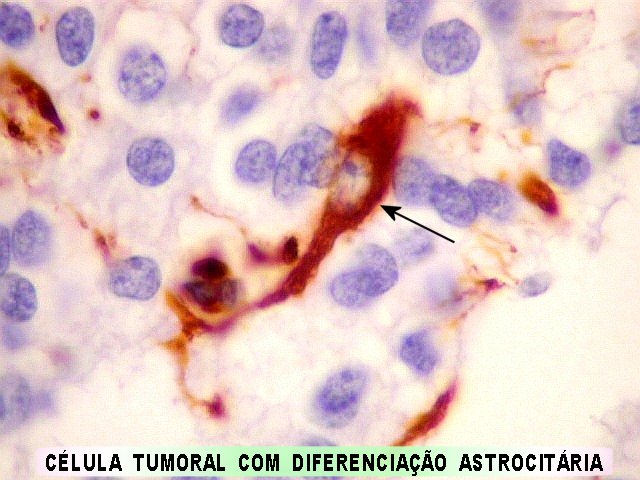
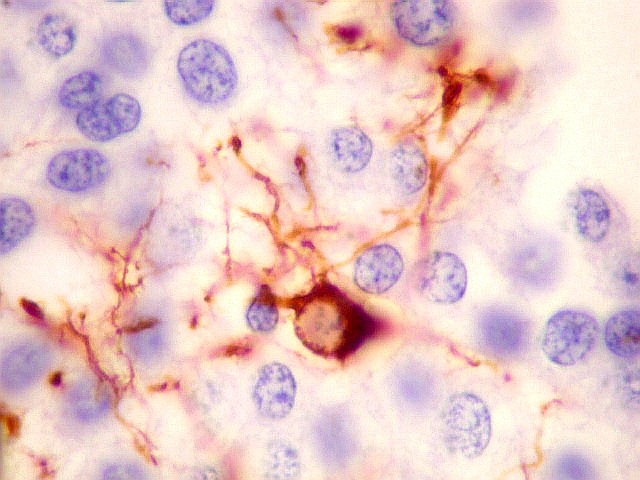
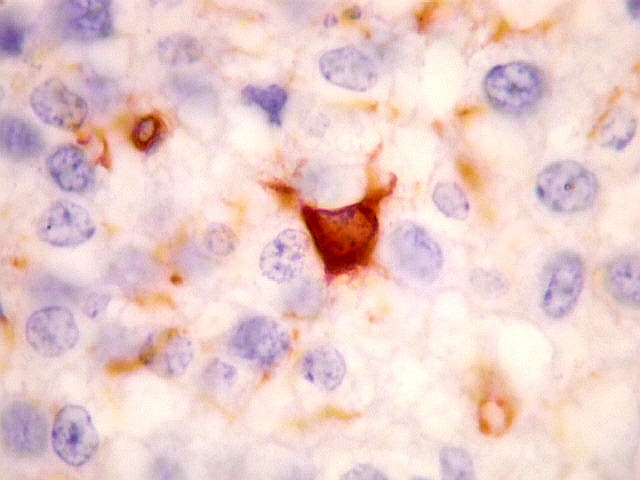
| VIM. Células neoplásicas com diferenciação astrocitária. Aqui vale o já dito com GFAP - as células marcadas não parecem astrócitos pré-existentes. Os núcleos são semelhantes em tamanho aos das demais células neoplásicas e os prolongamentos citoplasmáticos são relativamente curtos e pouco ramificados. Comparar com astrócitos reativos na periferia do tumor, abaixo. | |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
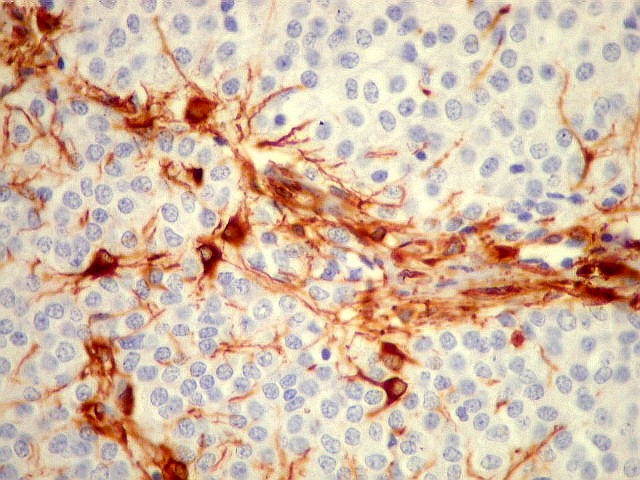
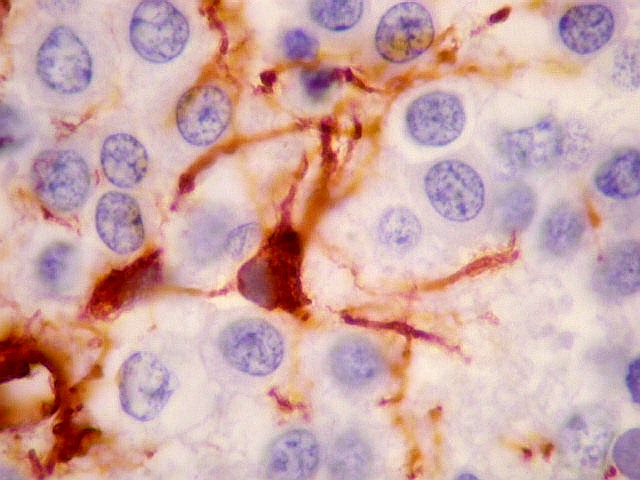
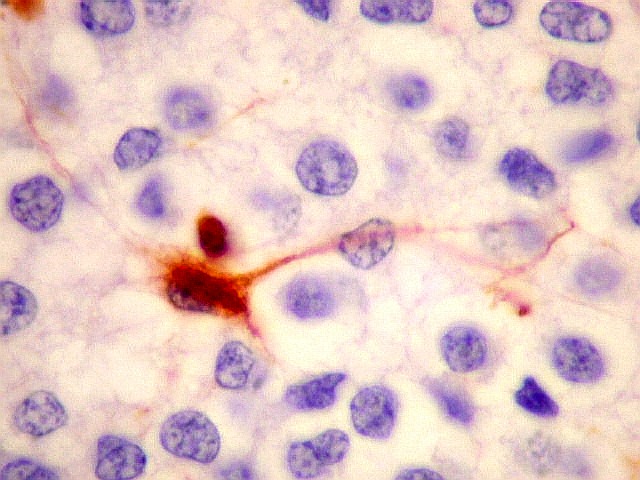
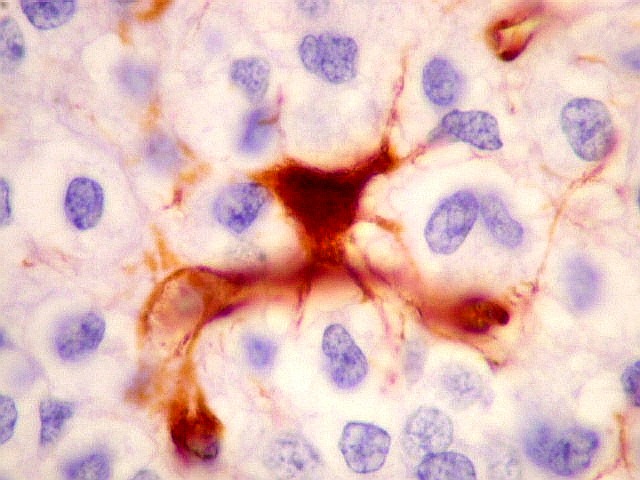
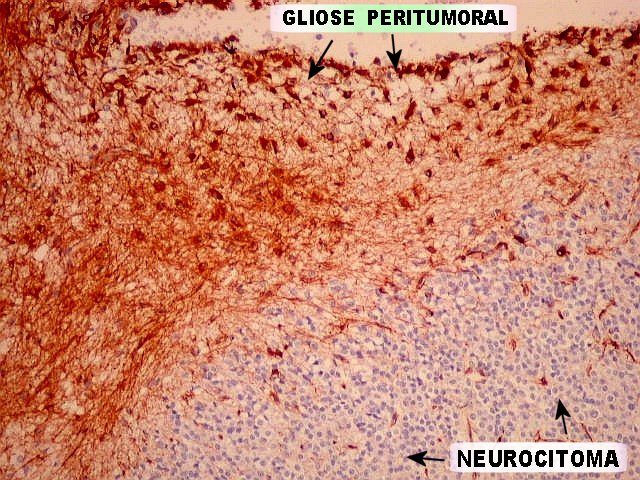
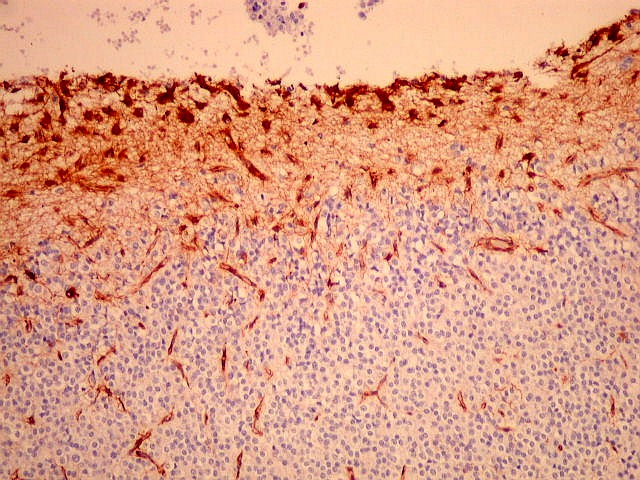
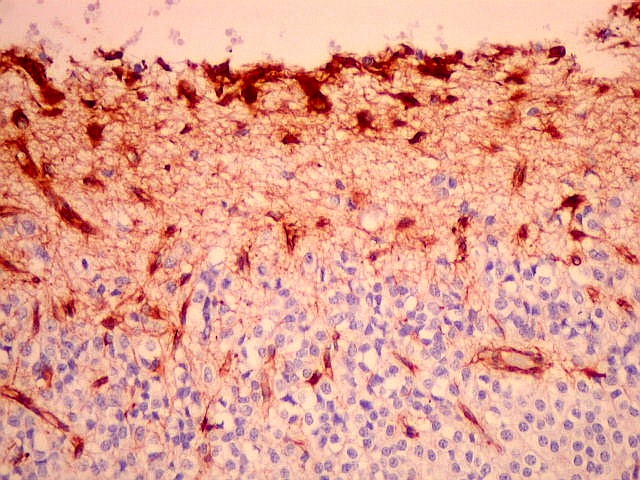
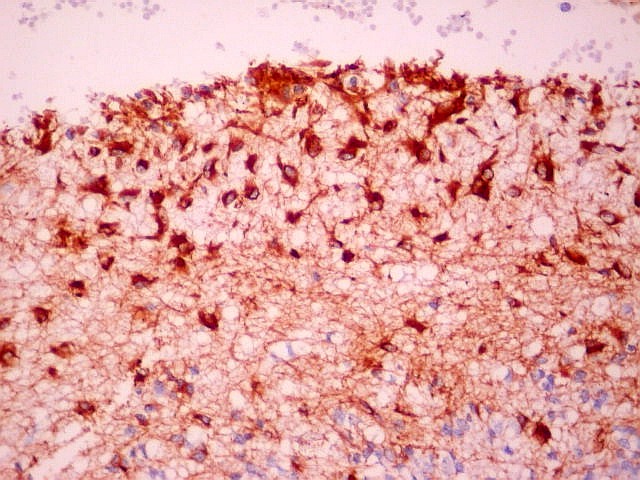
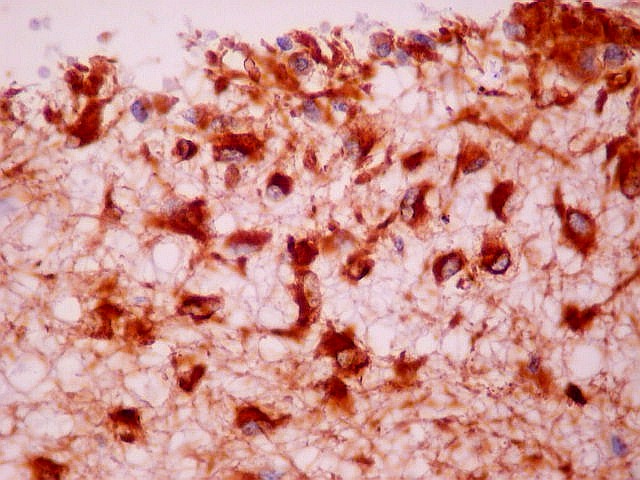
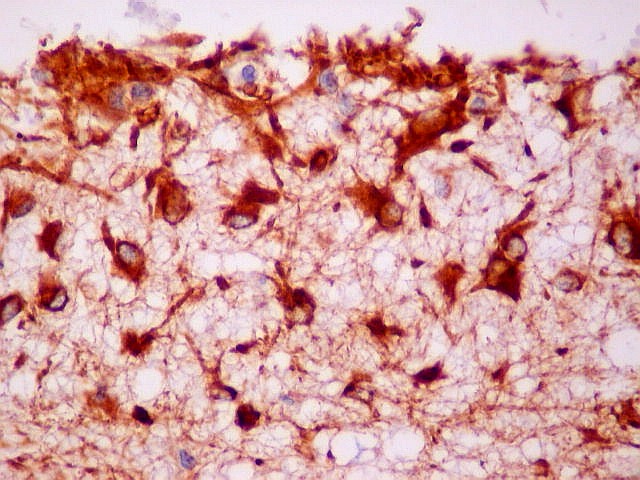
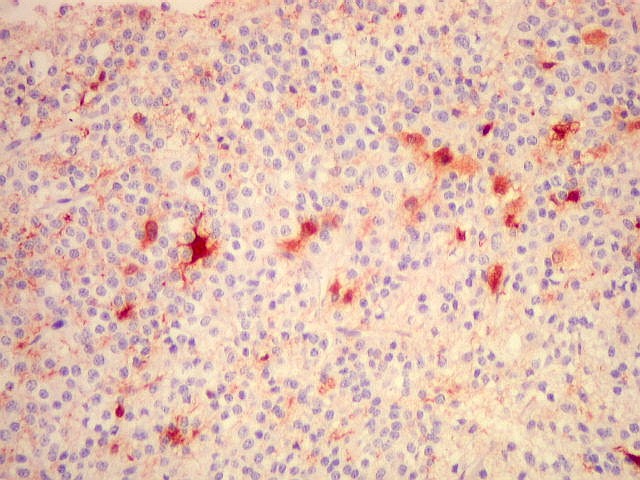
| VIM. Gliose na periferia do tumor. Aqui os astrócitos são maiores, densamente agrupados e têm citoplasma mais abundante, correspondendo a verdadeiros astrócitos gemistocíticos (formas reacionais ao tumor). | |
 |
 |
 |
 |
 |
 |
 |
 |
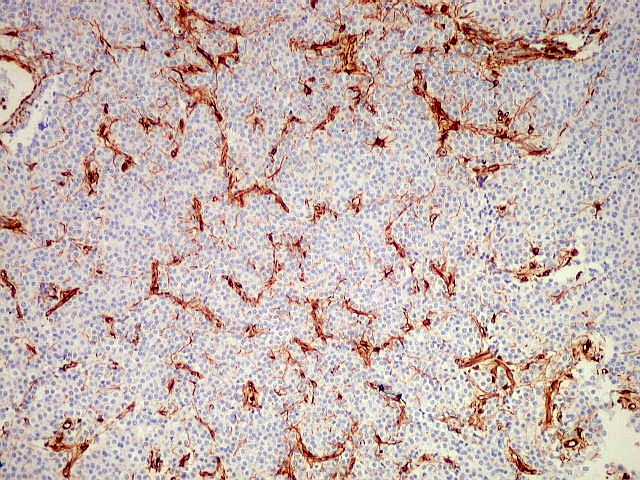
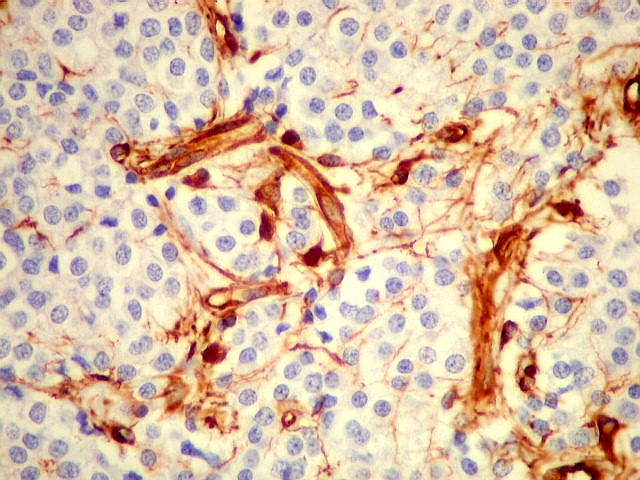
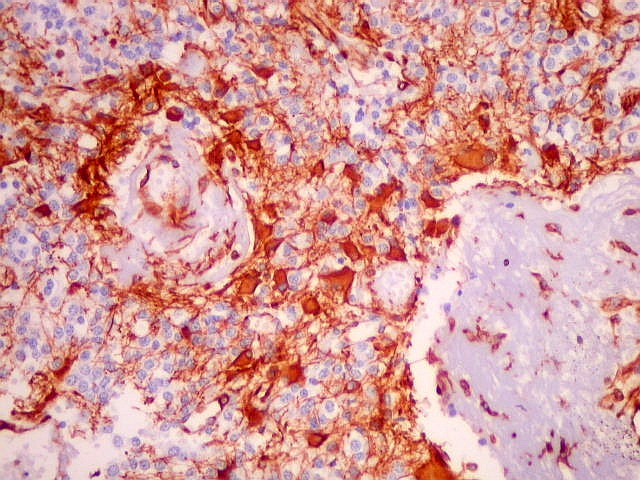
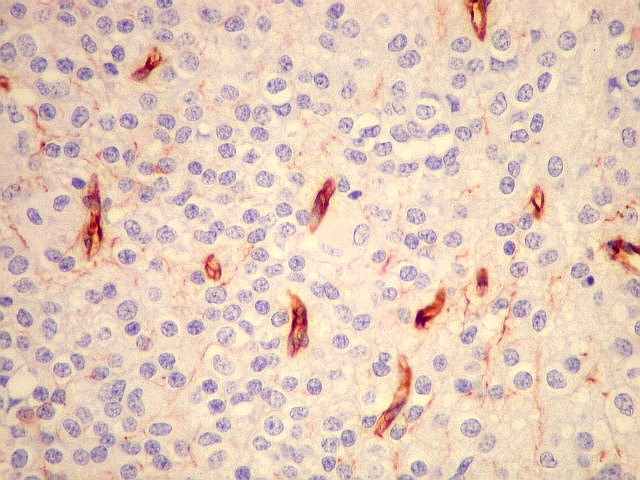
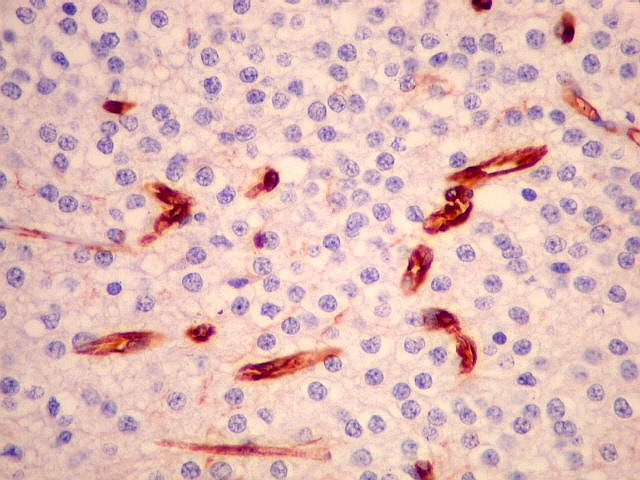
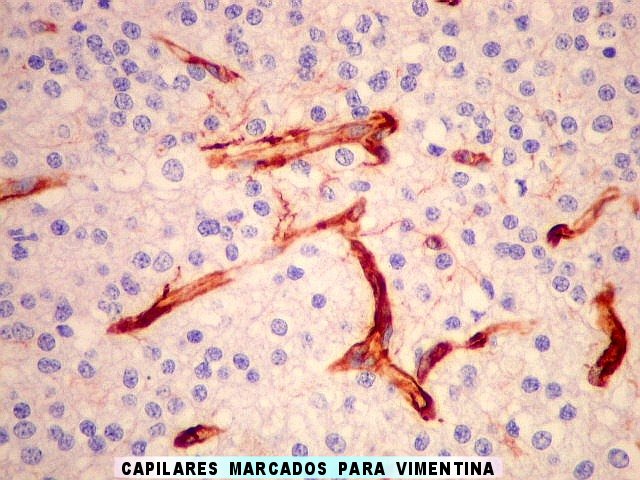
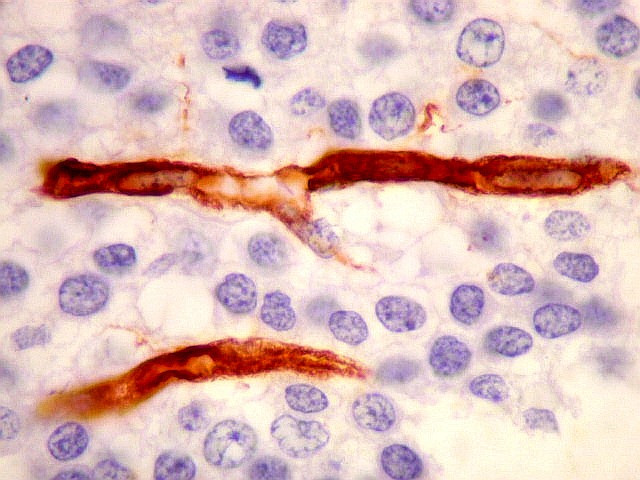
| VIM. Capilares. A vimentina é positiva em vasos tumorais, inclusive na rede capilar, sendo um dos anticorpos de escolha para demonstrar a vascularização da neoplasia. Aqui, os capilares são delicados, regularmente espaçados e não mostram proliferação endotelial, como em HE. Com CD34, positivo em células endoteliais, o resultado é superponível. | |
 |
 |
 |
 |
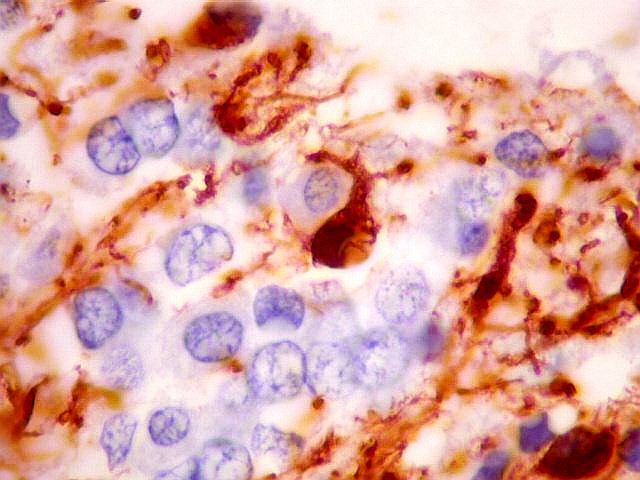
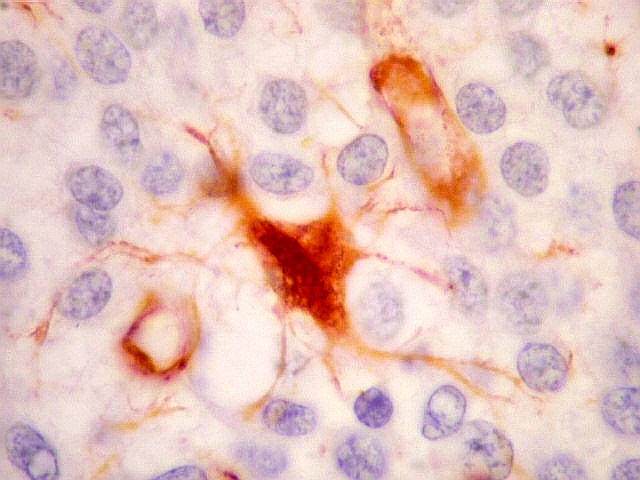
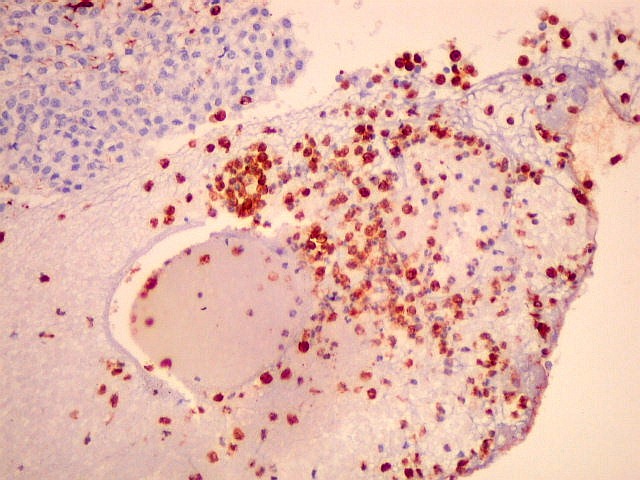
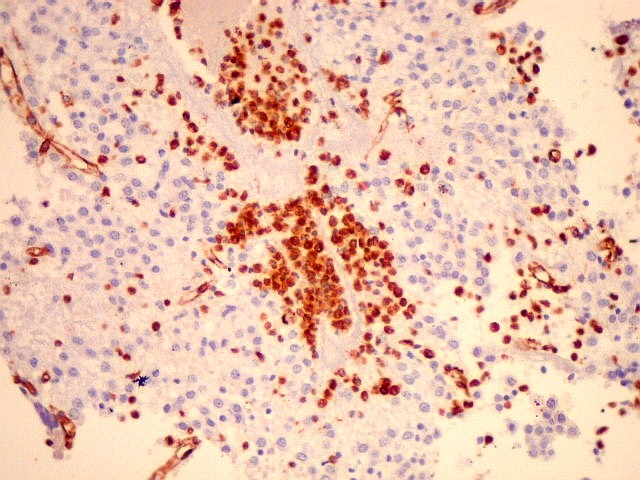
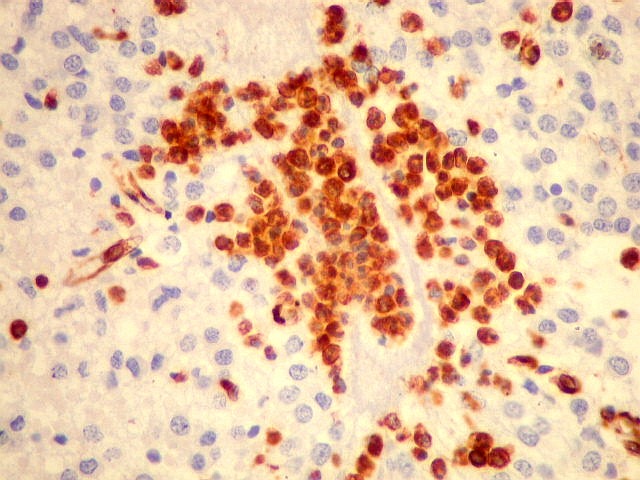
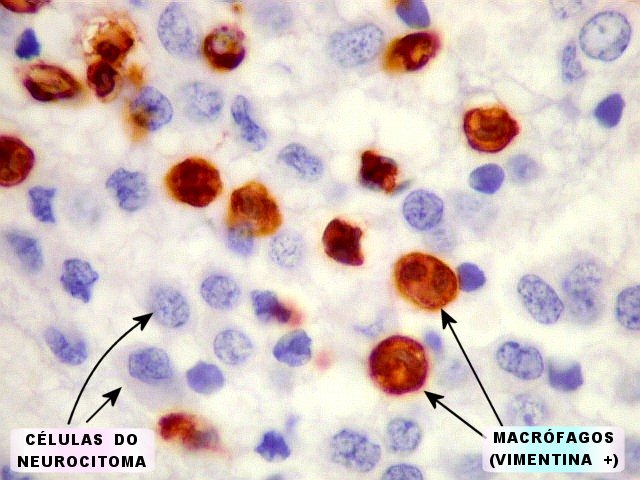
| VIM. Macrófagos. Macrófagos estão presentes em localização perivascular e entre as células neoplásicas, provavelmente fagocitando restos celulares. São fortemente positivos no citoplasma, ficando o núcleo mascarado pelo produto da reação. | |
 |
|
 |
 |
 |
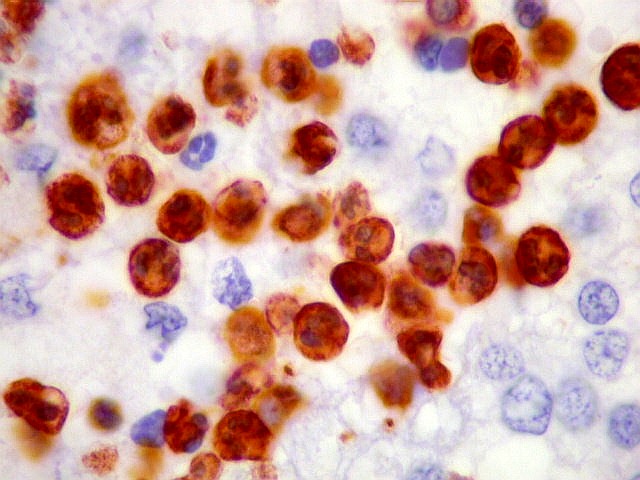 |
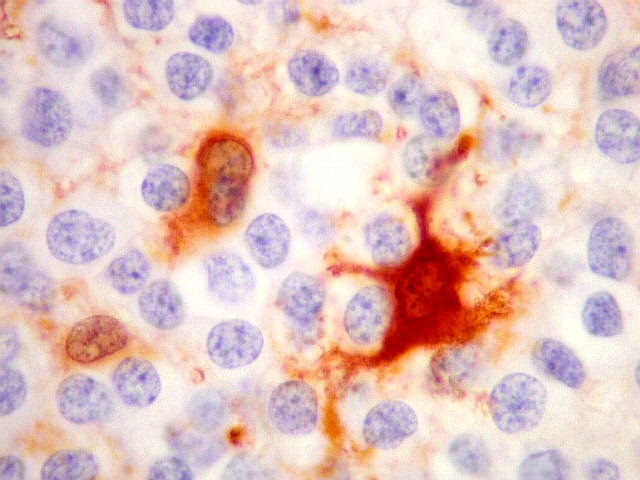
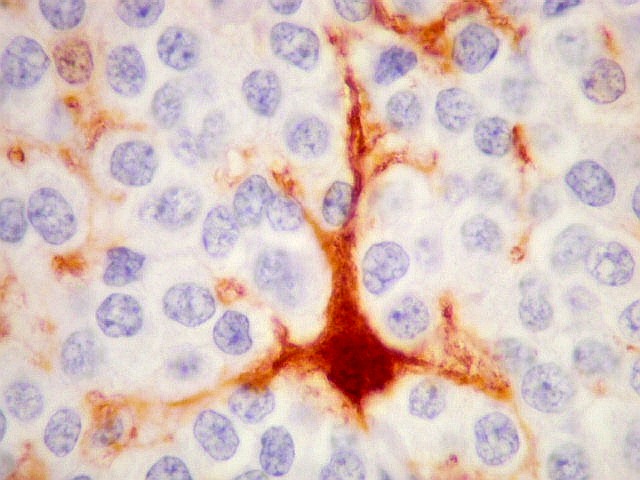
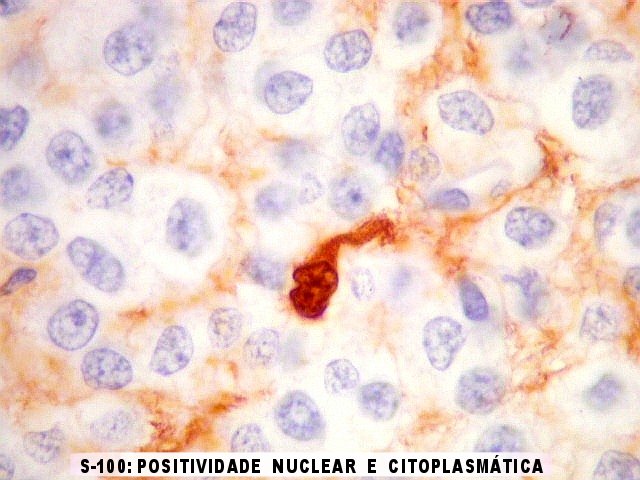
| S-100. Positividade nuclear e citoplasmática em parte das células neoplásicas, provavelmente as com diferenciação astrocitária, como com GFAP e VIM. | |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
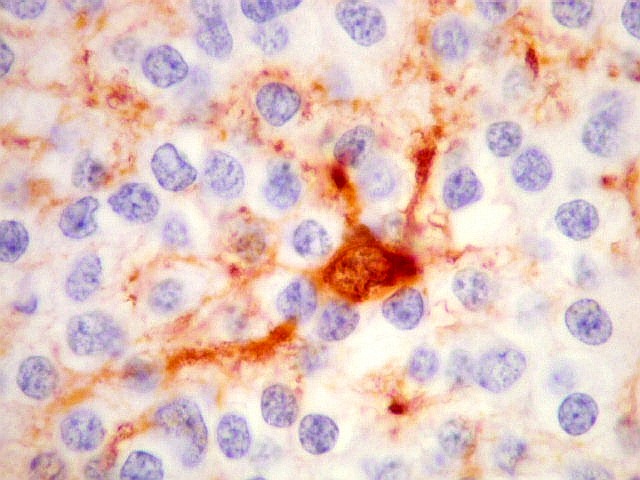
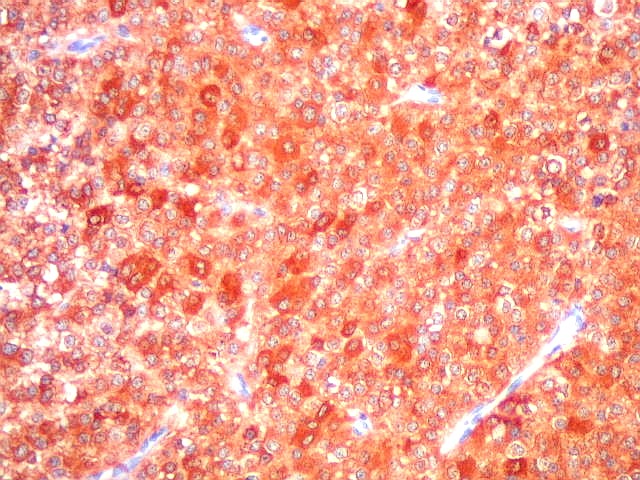
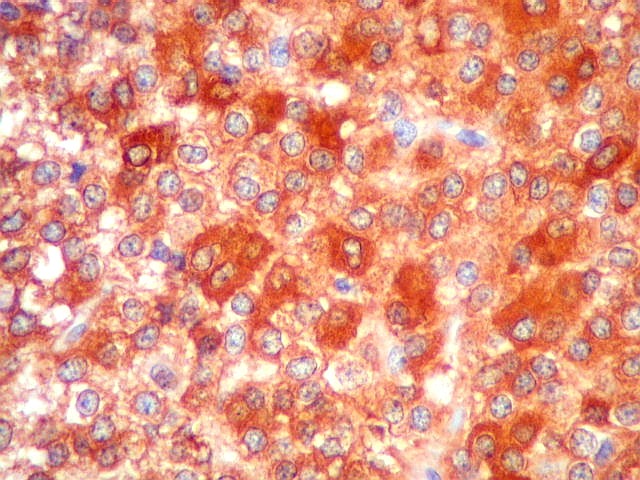
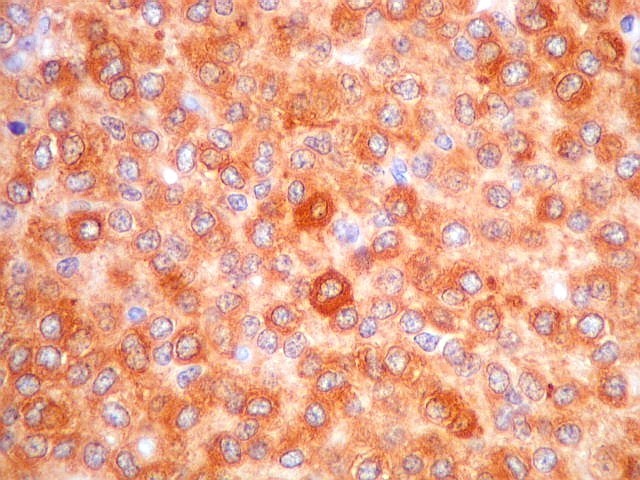
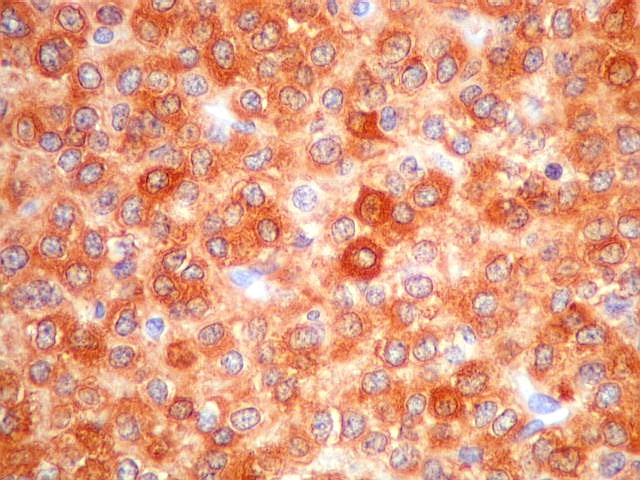
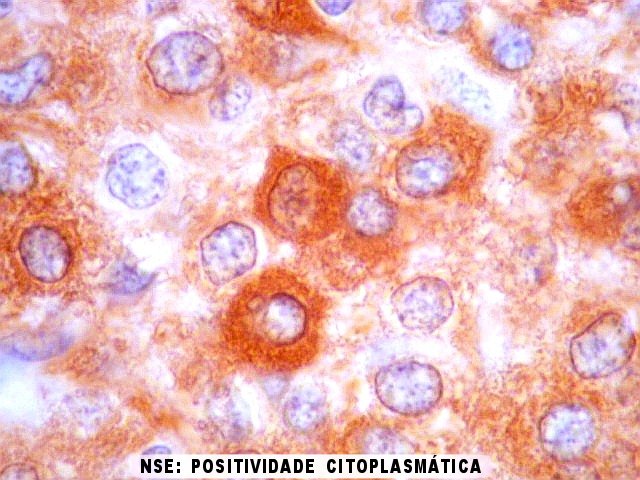
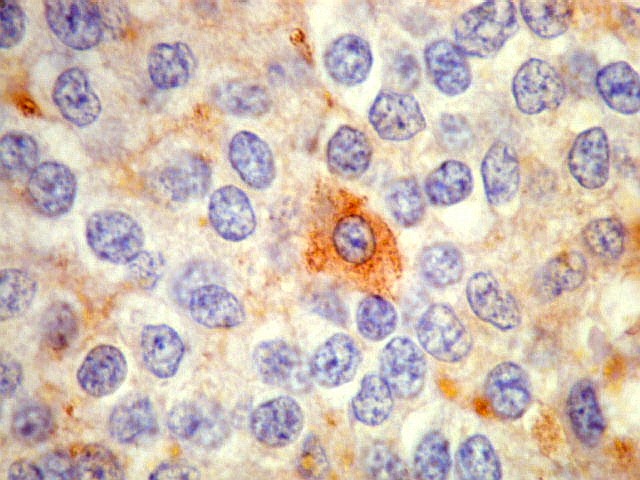
| NSE. Positividade citoplasmática forte em grande parte das células neoplásicas, sugerindo diferenciação neuronal. Contudo, NSE (enolase neurônio-específica) é, na realidade, pouco específica, sendo positiva também em gliomas, como oligodendrogliomas e ependimomas. | |
 |
 |
 |
 |
 |
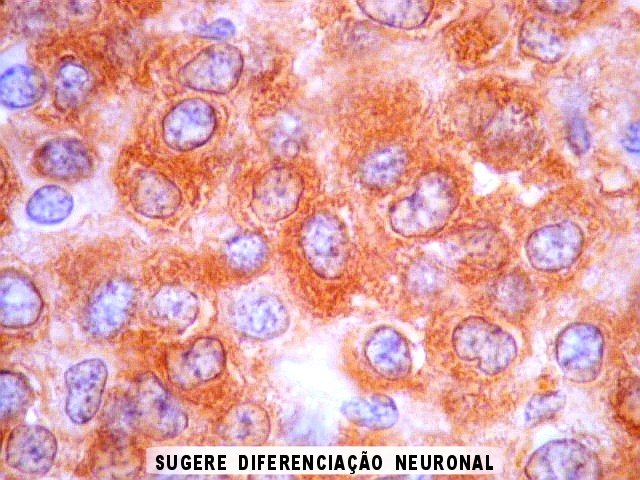 |
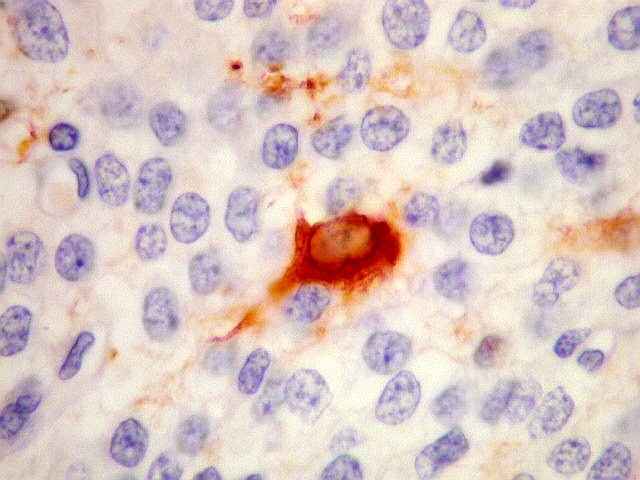
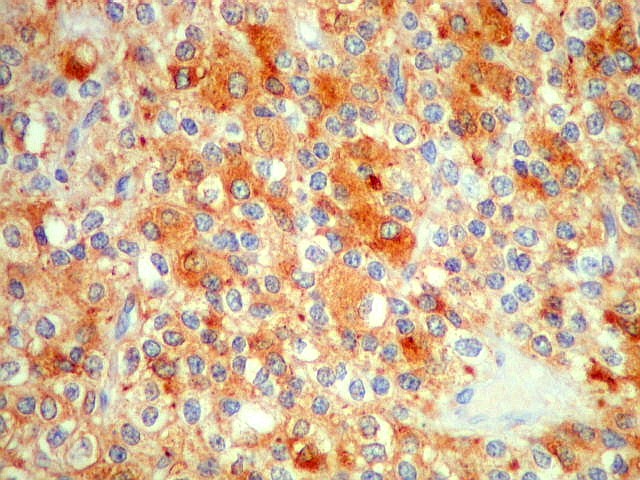
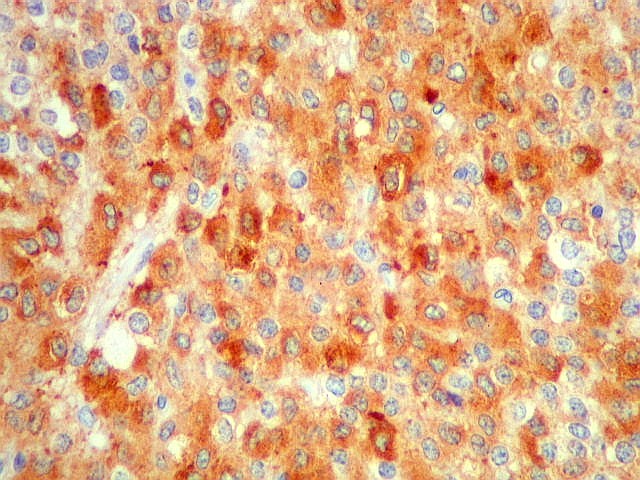
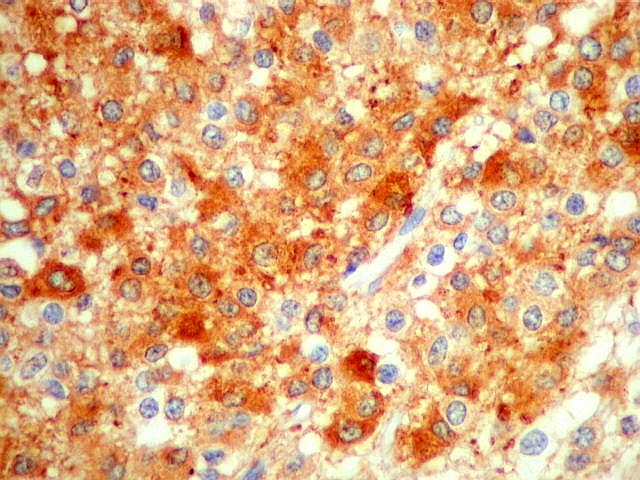
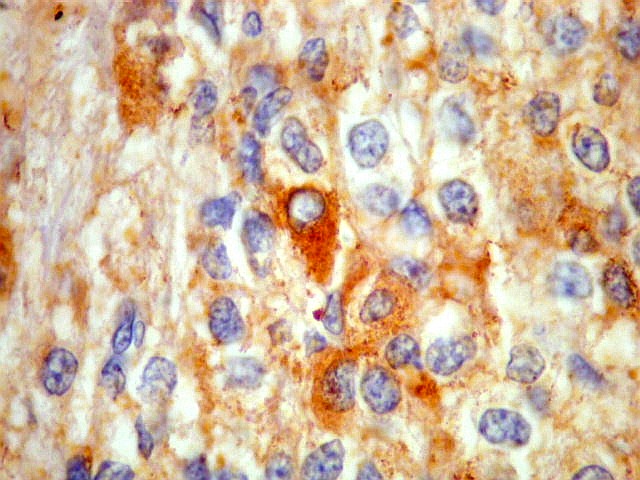
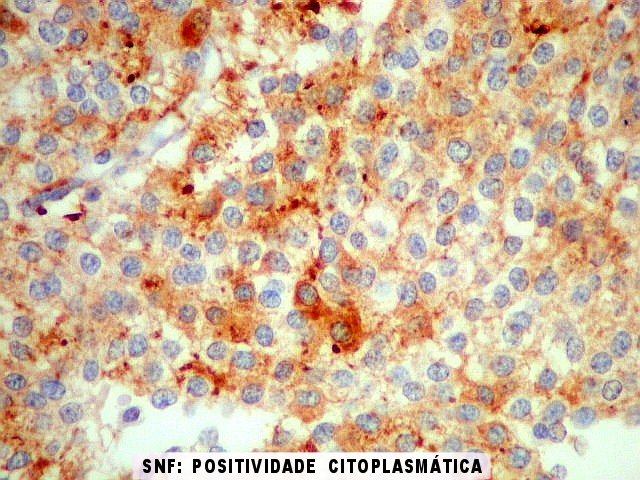
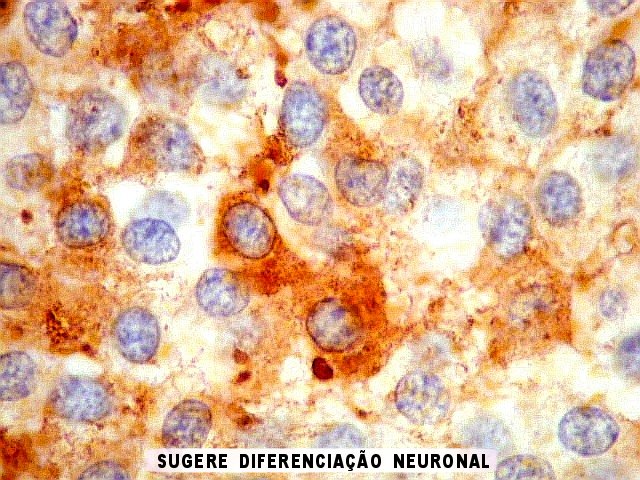
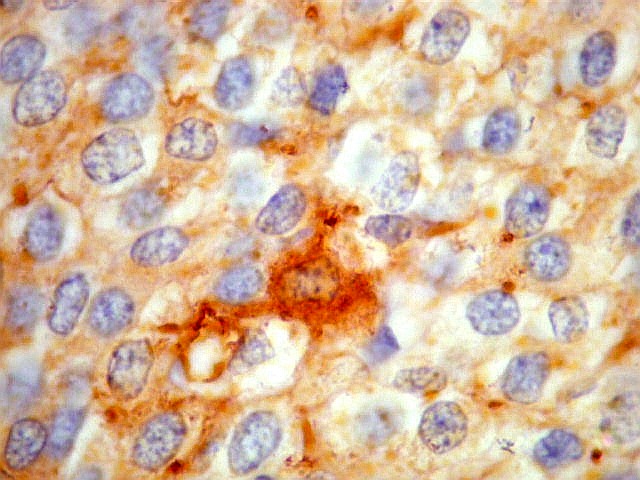
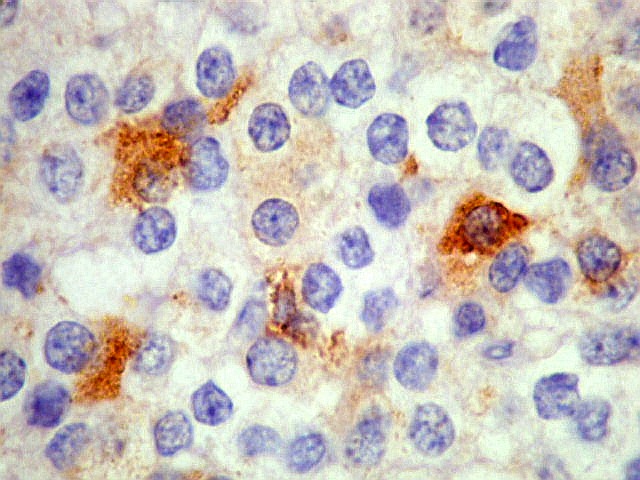
| SNF. Positividade citoplasmática forte em grande parte das células neoplásicas, que contrastam com células não reativas. Ao contrário de NSE, SNF e CGR apresentam um grau maior de especificidade para linhagem neuronal. | |
 |
 |
 |
 |
 |
 |
 |
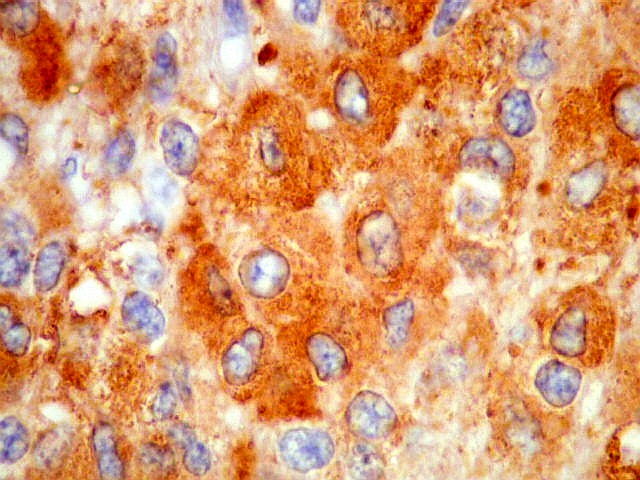 |
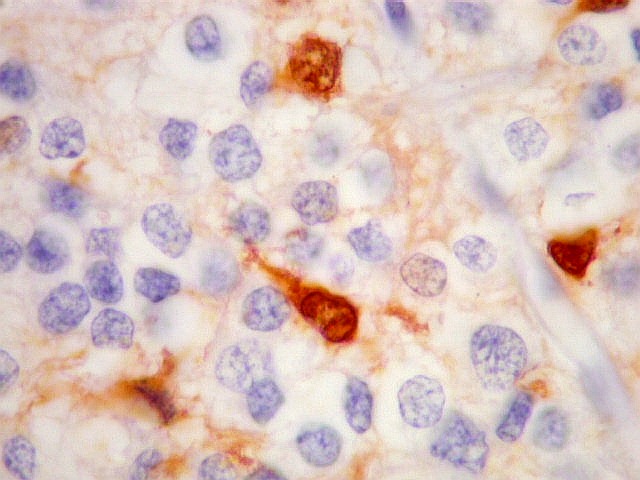
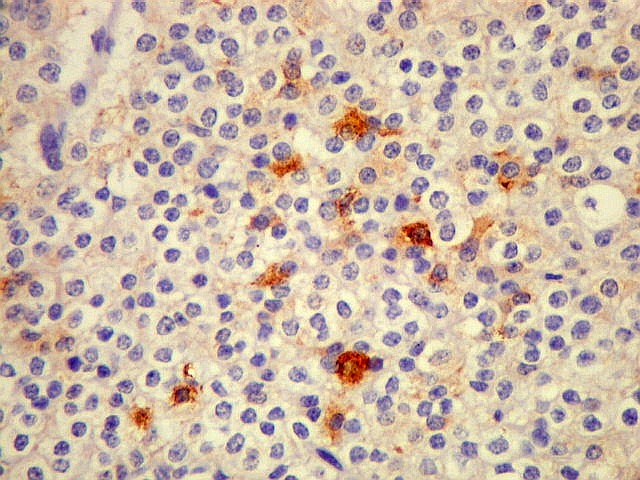
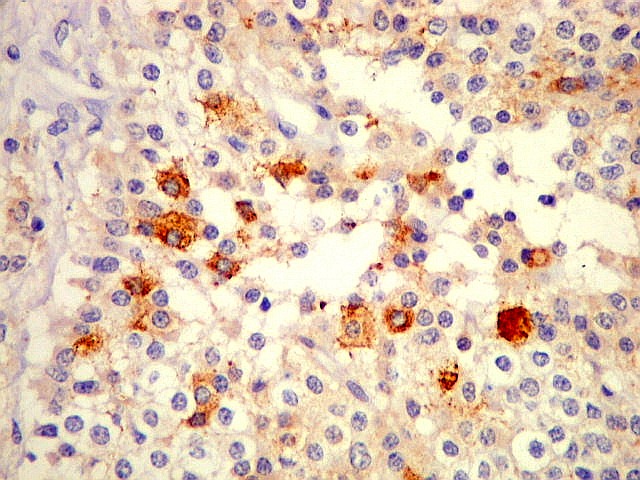
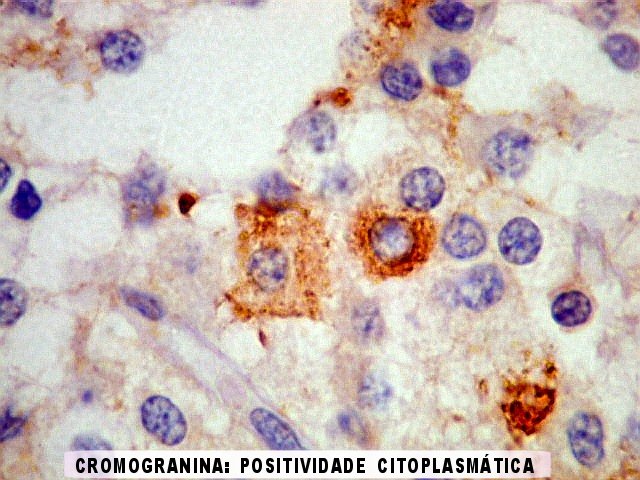
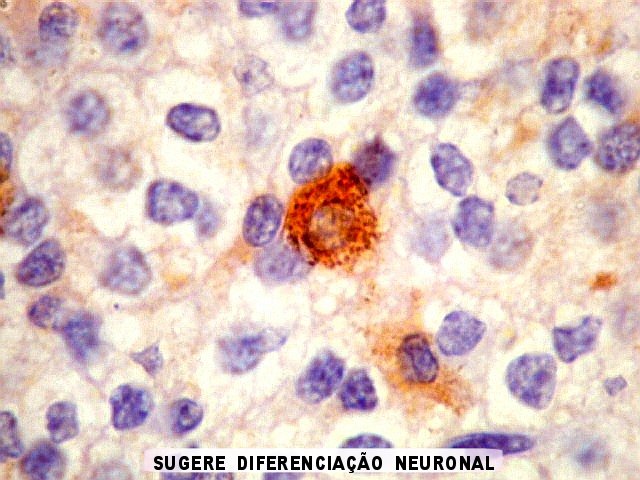
| CGR. Menos células reativas que com NSE ou SNF, mas as que reagem são fortemente positivas e destacam-se das demais. A reatividade é citoplasmática. | |
 |
 |
 |
 |
 |
 |
| NF. Negativo em toda a amostra. A negatividade indica que as células neoplásicas, embora apresentando diferenciação neuronal, como comprovado pela reatividade para SNF e CGR, estão num estágio intermediário. Só células com grau avançado de diferenciação para neurônios (chamadas células ganglionares, com o núcleo típico com cromatina frouxa, nucléolo evidente e citoplasma abundante com corpúsculos de Nissl) costumam ser positivas para neurofilamento. | |
 |
 |
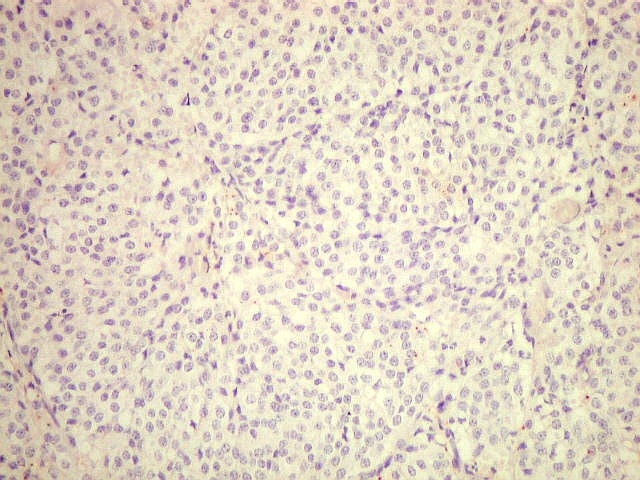
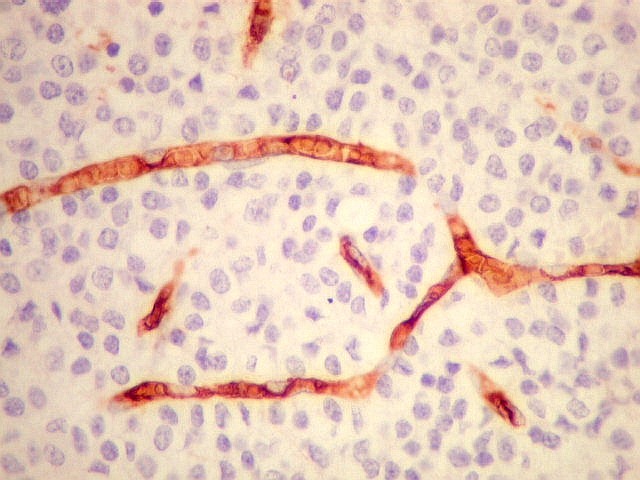
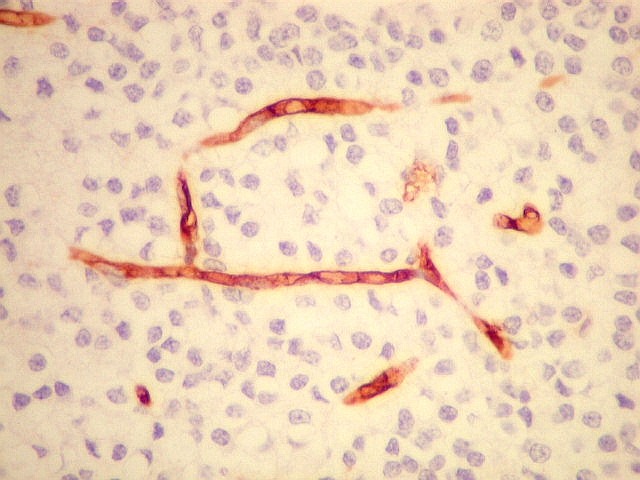
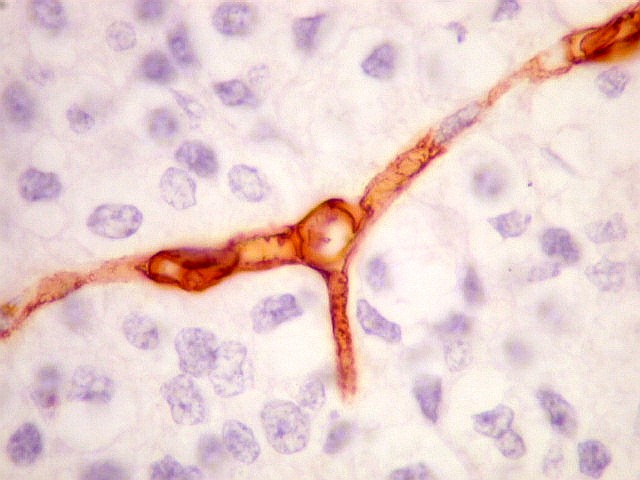
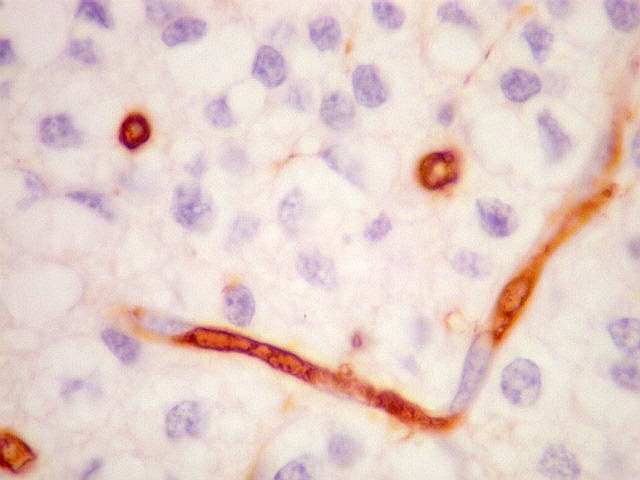
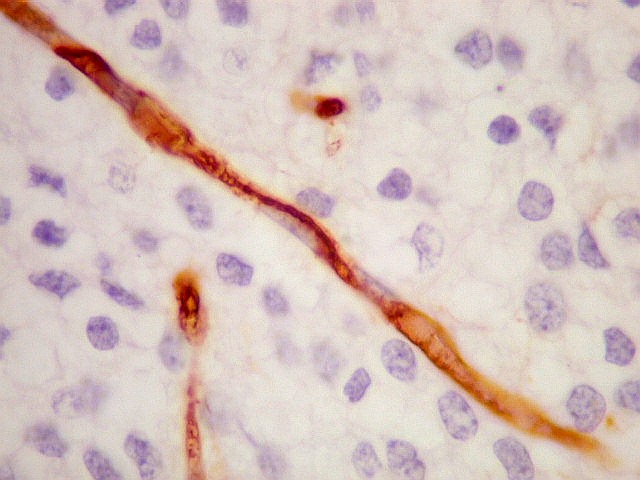
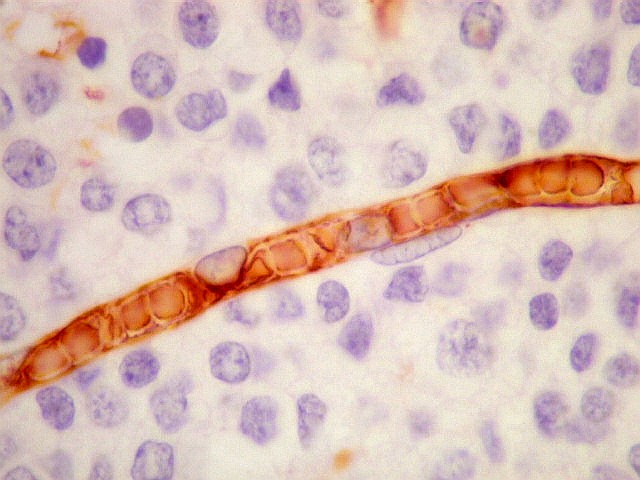
| CD34. Positivo nas células endoteliais, demonstra com elegância a rede capilar. Notar que os capilares são finos, com ramificação dicotômica, sem espessamentos sugestivos de proliferação vascular. O aspecto é muito semelhante ao encontrado em oligodendrogliomas, e classicamente comparado a 'tela de galinheiro'. | |
 |
 |
 |
 |
 |
 |
 |
 |
 |
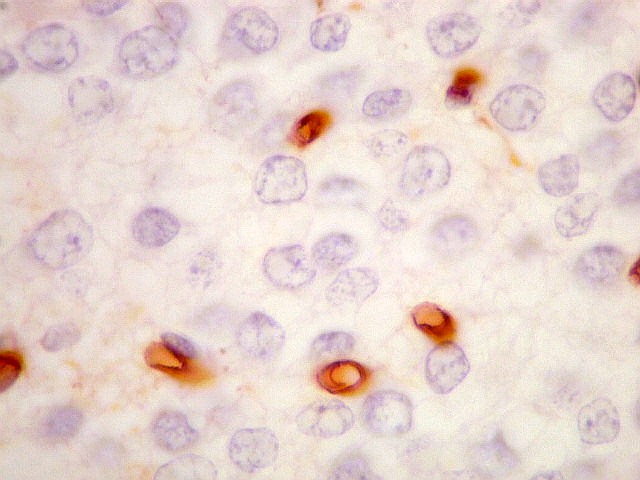 |
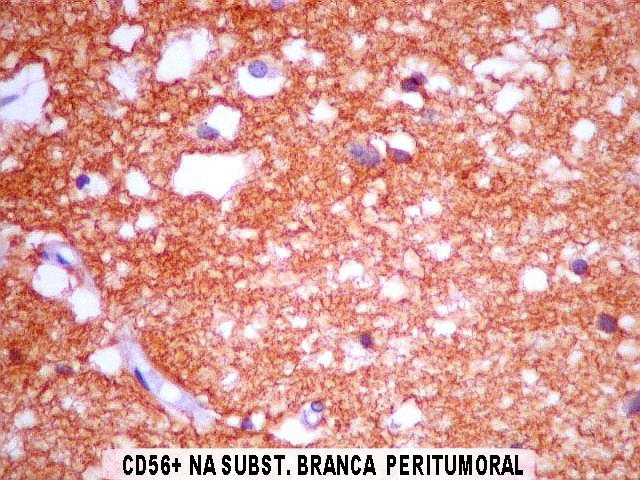
| CD56. Esta molécula de adesão intercelular (NCAM ou neural cell adhesion molecule) é difusamente positiva no neurocitoma a nível das membranas celulares. Porém, é inespecífico, sem valor para diferenciá-lo de outros tumores, como o oligodendroglioma. | |
 |
 |
 |
 |
 |
 |
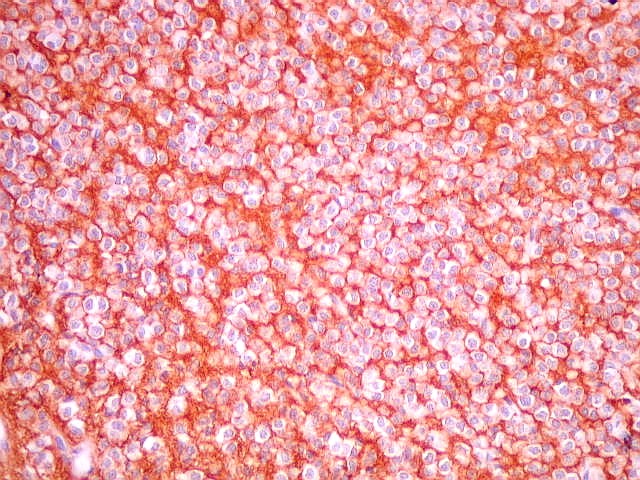
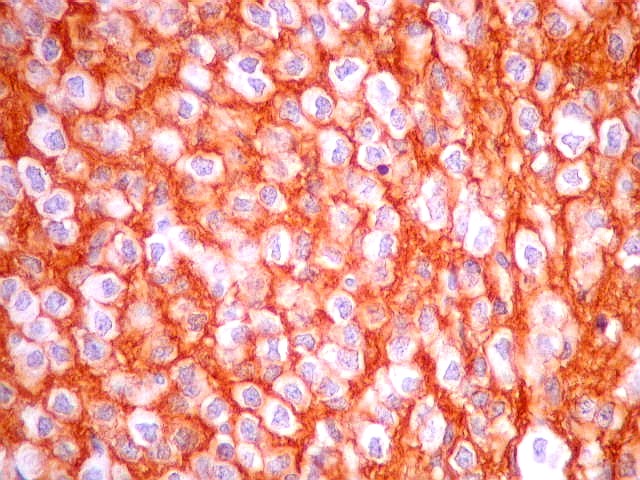
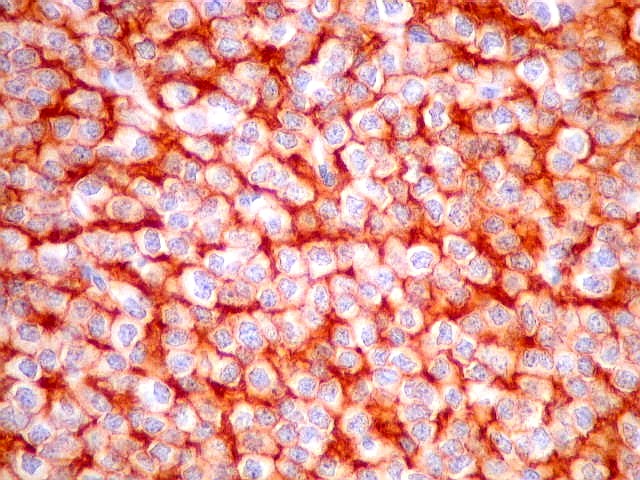
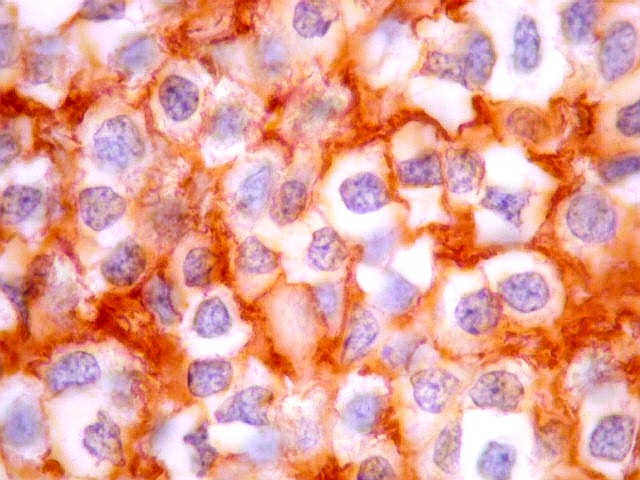
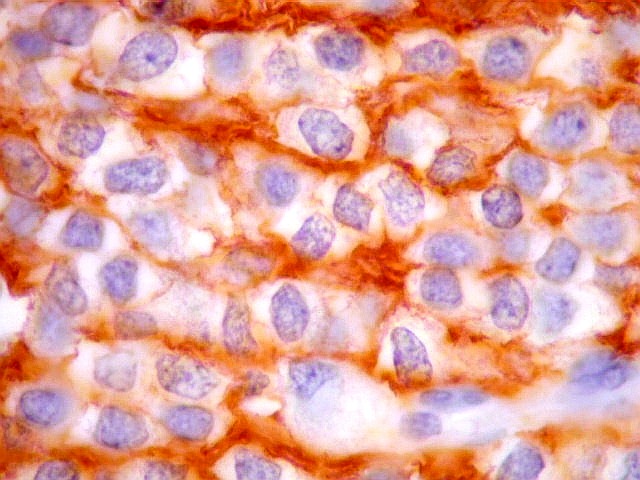
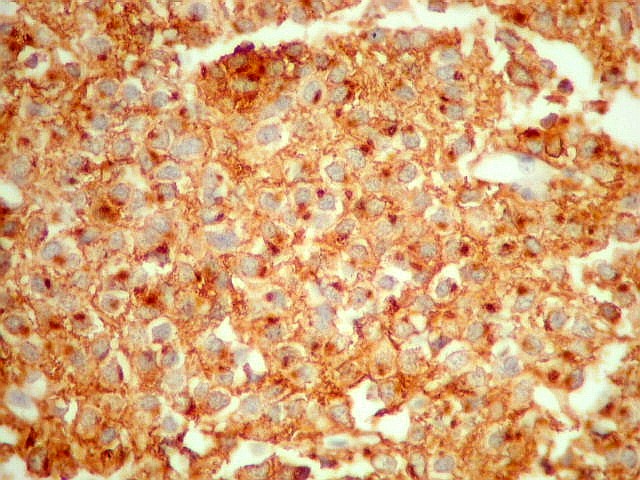
| CD57. Esta outra molécula de adesão intercelular, também chamada Leu-7, é positiva difusamente, freqüentemente em padrão dot (formando uma pequena área fortemente positiva no citoplasma das células neoplásicas). | |
 |
 |
 |
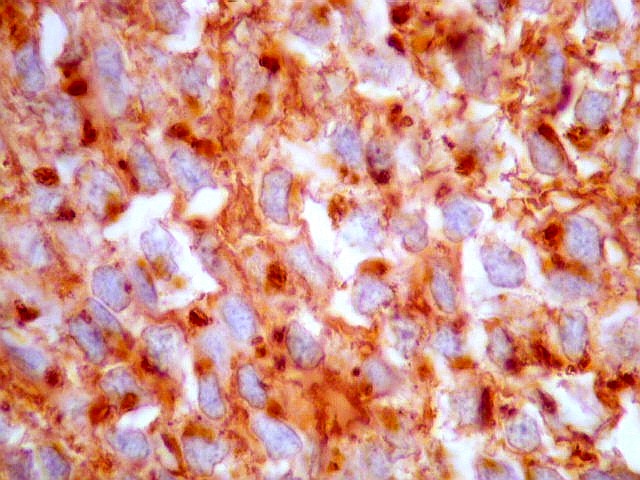 |
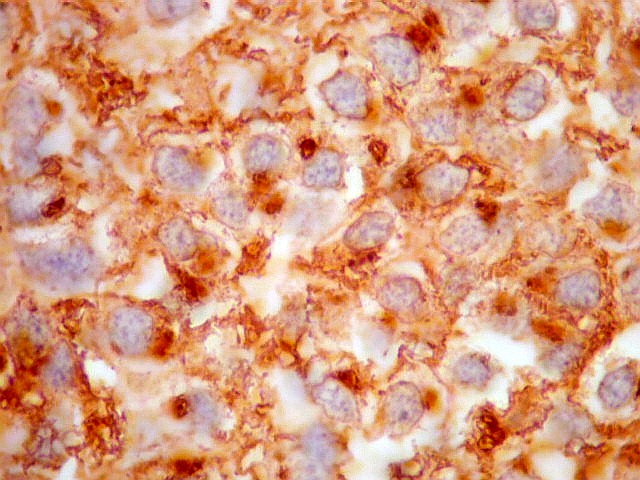
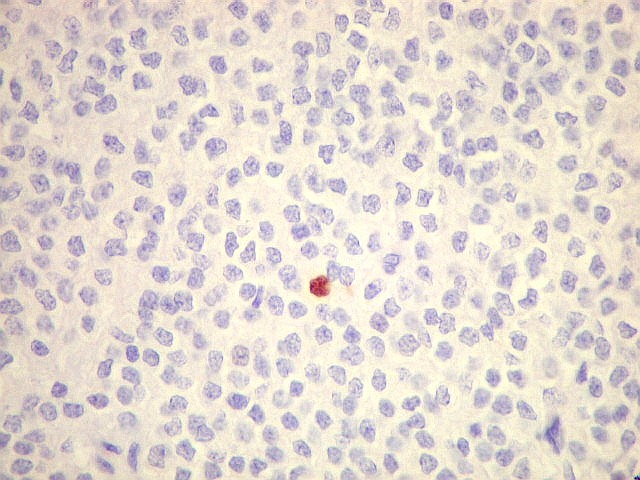
| KI-67. Positivo em poucos núcleos, menos que 1%, indicando baixa proliferação celular. | |
 |
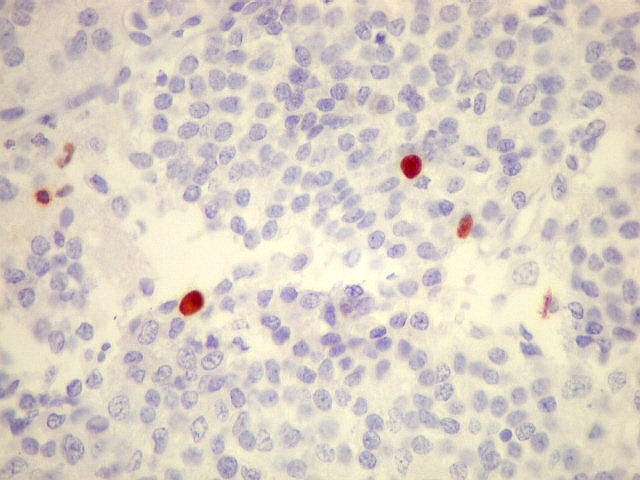 |
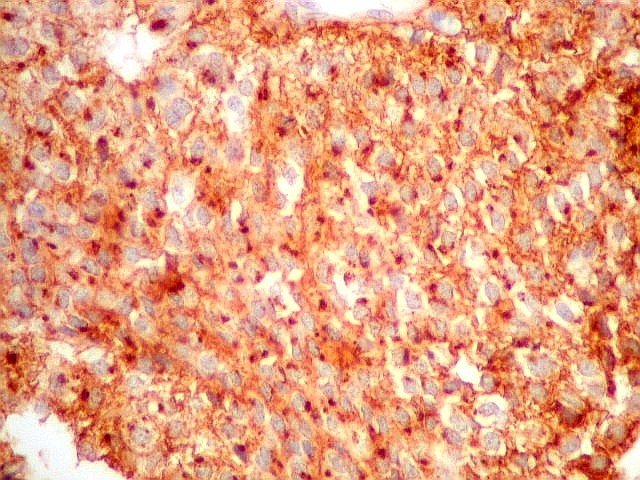
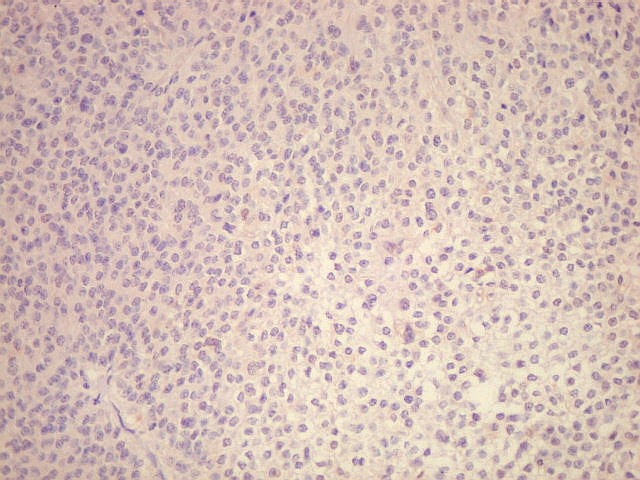
| p53. Negativo em toda a amostra, sugerindo que uma mutação do gene supressor tumoral p53 não deve ter papel importante na gênese do neurocitoma central. | |
 |
 |
|
Neurocitoma central Definição. Tumor constituído por células redondas uniformes, com diferenciação neuronal demonstrável por imunohistoquímica e/ou microscopia eletrônica, tipicamente situado nos ventrículos laterais na região do foramen de Monro, afetando adultos jovens, e com bom prognóstico. Histologicamente imita oligodendrogliomas. Apresenta grau de malignidade II segundo a OMS. Histórico. Originalmente descrito por Hassoun e colaboradores (1982). O termo deve ser restrito a tumores situados nos ventrículos supratentoriais. Incidência. Em 3 séries cirúrgicas, variou entre 0,25 e 0,5% de todos tumores intracranianos. A idade quando da manifestação clínica variou de 8 dias a 67 anos (média 29 anos). 46% foram diagnosticados na 3ª década e 72% entre as idades de 20 e 40 anos. Ambos sexos são igualmente afetados. Localização. Ventrículos laterais e/ou IIIº ventrículo. A posição mais comum é na porção anterior de um dos ventrículos laterais (50%), com certa preferência pelo E, seguido por uma extensão combinada aos ventrículos laterais e IIIº ventrículo (15%), ventrículos laterais bilateralmente (13%) e IIIº ventrículo apenas (3%). Neurocitomas pequenos foram descritos no forâmen de Monro, septo pelúcido, corpo caloso, hipotálamo e tálamo. Em 2 casos foi relatada disseminação liquórica. Clínica. A maioria dos pacientes apresenta hipertensão intracraniana de duração relativamente curta (média 3,2 meses). Ocasionalmente: déficits visuais ou mentais, distúrbios hormonais: amenorréia, gigantismo, hipersecreção de vasopressina ou de hormônio de crescimento pelo tumor. Imagem.
Macroscopia. Homogêneos, acinzentados, friáveis, com variável calcificação. Microscopia. Geralmente, aspecto semelhante a oligodendroglioma. Rosetas de Homer Wright e células ganglionares são raras. Capilares delicados. Calcificações em metade dos casos, geralmente distribuídas por todo o tumor. Raramente, anaplasia, mitoses, proliferação vascular e necrose. Diagnóstico diferencial: oligodendroglioma, ependimoma, pineocitoma. Imunohistoquímica. Sinaptofisina é o marcador mais confiável. Contudo, em biópsias pequenas, neurópilo e neurônios preexistentes podem complicar a interpretação. Fixação prolongada pode causar negatividade da sinaptofisina. Outros marcadores neuronais expressos são NSE (sempre positiva), b-tubulina classe III, proteína tau, MAP-2 e calcineurina. Cromogranina e neurofilamento estão geralmente ausentes. GFAP quase sempre ausente na maior parte das células, mas pode marcar células individuais. Parte dos autores interpreta isto como astrócitos pré-existentes, outros como indício de diferenciação glial. Microscopia eletrônica. Processos celulares finos e entrelaçados, com características de prolongamentos de neurônios: microtúbulos, filamentos intermediários, vesículas claras e vesículas de centro denso. Sinapses podem estar presentes, mas são raras e não necessárias para o diagnóstico. Proliferação. Quando medida por Ki-67, geralmente menor que 2%. Tumores com índice maior que 2% freqüentemente mostraram proliferação vascular e tiveram um intervalo significativamente mais curto antes da recidiva. Como o prognóstico, mesmo assim, é muito melhor que o dos gliomas, fala-se em neurocitoma atípico, e não anaplásico. Citometria de fluxo revelou diploidia em 10/10 casos. Genética. Em 12 casos não foi detectada mutação de p53. Histogênese. Admite-se origem em remanescentes das células da matriz periventricular. Ainda não se sabe se de células já comprometidas com o fenótipo neuronal, ou de células progenitoras bipotenciais (para neurônio e astrócito). É relatada co-expressão de GFAP e sinaptofisina. Prognóstico. Geralmente benigno. O tratamento de escolha é remoção cirúrgica total. Pacientes que não receberam radioterapia adjuvante tiveram sobrevida longa sem recidiva, mas a maioria dos pacientes é irradiada. Não se sabe se isto é realmente necessário. Há alguns casos com histologia benigna e baixos índices proliferativos que tiveram recidivas e má evolução.
|
| Para exames de imagem deste tumor, clique » |  |
| Sobre o neurocitoma central |
| Neuropatologia
- Graduação |
Neuropatologia -
Casos Complementares |
Neuroimagem
- Graduação |
Neuroimagem -
Casos Complementares |
Correlação
Neuropatologia - Neuroimagem |
|
|
|
|